
Abstract
Increasing evidence showed that altered histone deacetylases (HDACs) are involved in, and exert cell type specific roles in sarcomagenesis. Here we reviewed expression and roles of HDACs in several types of human sarcomas, providing a basic guideline for personalized therapy. In sarcomas, overexpression of HDACs was common, including a prevalence in uterine sarcomas. Class I HDAC1–3, especially HDAC1-2, were upregulated in most sarcomas and were often associated with poor prognosis. Class I HDACs inhibit tumor suppressor expression and lineage differentiation, but maintain chromatin integrity and oncofusion protein stability. Class II HDACs have specific functions in cellular transformation, long telomeres maintenance, drug resistance, and cytoskeletal organization, and act as negative predictors in some sarcomas; for example, HDAC4 for leiomyosarcoma (LMS) and endometrial stromal sarcoma (ESS), HDAC5 for uterine LMS, HDAC6 for ESS, chondrosarcoma (CHS) and undifferentiated endometrial sarcoma. Moreover, some HDACs play dual, cell-context or cellular localization-dependent roles. HDAC2 and HDAC5 could promote or suppress osteosarcoma growth. HDAC4 acts as a tumor suppressor in CHS but as an oncogene in other sarcomas. Moreover, HDAC4 is the targets of several microRNAs in osteosarcoma. Cytoplasmic HDAC6 increases self-renewal and cell migration, compared with nuclear HDAC6 enhancing EWSR1-FLI1 transcription. Thus, the diverse expression and roles of HDACs in sarcoma pathogenesis will be a solid foundation to guide personalized therapeutic application of HDAC modulators in sarcomas.
Introduction
Sarcoma is a heterogeneous group of mesenchymal origin tumors, comprising over 60 different subtypes arising in soft tissues and bones. It is rare (accounting for <1% of cancers) in adults, but it occurs in 21% of pediatric malignancies. 1 The local disease, after a complete surgical resection, generally has a good prognosis. However, due to low response rates and resistance to conventional chemotherapy, the overall 5-year survival rate is only 50–60% in soft tissue sarcoma (STS) patients of all stages 2 and even as low as 20–30% in osteosarcoma and Ewing's sarcoma (EwS) patients with metastasis. 3 Thus, an alternative therapy is urgently needed.
Aberrant epigenetic regulation has been implicated in sarcoma occurrence at multiple levels. 4 Histone acetylation—the second most common epigenetic modification of chromatin—regulates the gene transcription through the dynamic balance of histone acetyltransferase (HAT) and histone deacetylase (HDAC). Acetylation of the lysine residue of histones by HAT results in relaxed chromosomal packing, allowing for transcription factors to access and initiate the gene transcription. Conversely, the removal of acetyl groups by HDAC promotes chromatin condensation, and is commonly associated with suppressed gene expression. HDACs also deacetylate various non-histone substrates to regulate gene transcription, DNA damage repair, cell division, signal transduction, and protein folding, etc. There are 18 human HDACs classified into four classes based on their structure, biochemical characteristics, and homology to yeast HDACs. Class I HDACs (consisting of HDAC1–3 and 8) are nuclear proteins and ubiquitously expressed. Class II HDACs are subdivided into IIA (HDAC 4, 5, 7, and 9) and IIB (HDAC 6 and 10). They are translocated between nucleus and cytoplasm, showing tissue-specific expression in muscle, cartilage, bone, immune system, and brain. Class III HDACs consist of SIRT1–7 proteins. Class IV HDAC contains only HDAC11. Class I, II, and IV HDACs are zinc2+-dependent, while class III SIRT is NAD+-dependent (see reviews by Li and Seto, Narita et al., and Seto and Yoshida5–7).
Class I and II HDACs play essential roles in the initiation and progression of sarcoma, being the main targets of the commercially available HDAC inhibitor (HDACi). Here, we review their expression profile (shown in Table 1) and molecular functions (shown in Table 2) in several types of human sarcomas, and provide an important foundation for an efficient personalized therapeutic application of HDAC modulators.
HDACs expression in sarcomas.
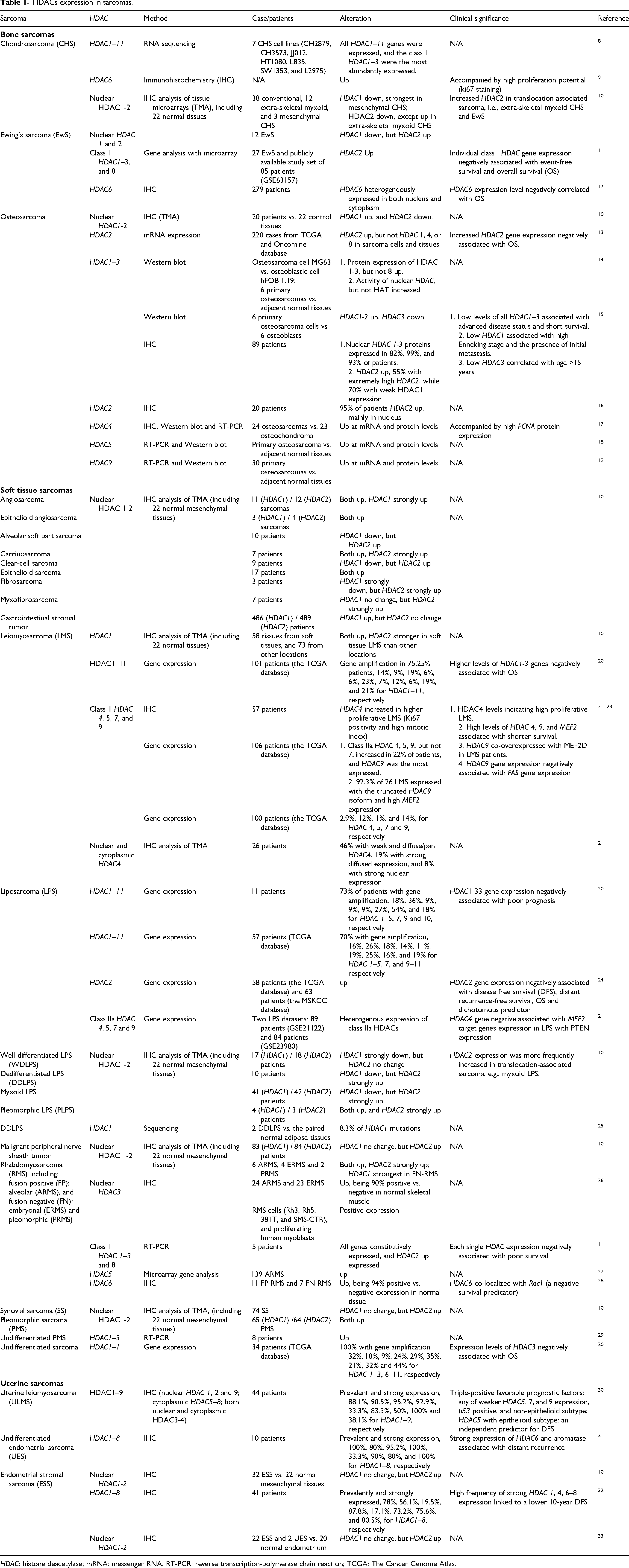
HDAC: histone deacetylase; mRNA: messenger RNA; RT-PCR: reverse transcription-polymerase chain reaction; TCGA: The Cancer Genome Atlas.
Roles of HDACs in sarcomas.


ESS: endometrial stromal sarcoma; HDAC: histone deacetylase; IL: interleukin; LMS: leiomyosarcoma; OS: osteosarcoma; PCNA: proliferating cell nuclear antigen; PD-L1: programmed death-ligand 1; SE: siRNA: small interfering RNA; STS: soft tissue sarcoma; TGFβ: transforming growth factor beta; ULMS: uterine leiomyosarcoma; VEGF: vascular endothelial growth factor; VPA: valproic acid.
Overview of HDAC expression in sarcomas
Alterations in HDAC genes were common in sarcomas, including messenger RNA (mRNA) high, amplification, deletion, and multiple mutations.20,71 An analysis of the sequencing data of 243 STS and bone sarcomas from The Cancer Genome Atlas (TCGA) database showed 41% of patients with gene copy amplification, deletion, and mutation of HDAC1–11. HDAC 2, 7, 9, and 11 were the most abundantly amplified in 3–5% of patients, HDAC 1 and 3 were amplified in 2% of patients. HDAC4 was the most frequently deleted: 7% of deletion versus 1% of amplification. 71 STSs presented the highest alterations, particularly mRNA high, in HDAC4 (30%) and other class IIa HDAC (almost 60%) among 38 tumor types (data from http://www.cbioportal.org). Interestingly, there were no gene mutations of class IIa HDAC in STSs versus the prominent mutations in melanoma, non-small cell lung cancer, and colorectal cancer. 71 Another analysis of the TCGA database reported HDAC1–11 gene amplifications in 76.65% of 257 STSs; that is, 91.67% of fibrosarcoma, 70% of liposarcoma (LPS), 100% of undifferentiated sarcoma, and 75.25% of leiomyosarcoma (LMS). 20
The protein levels of HDAC 1 and 2 were differently expressed in sarcomas. Analyzing 1196 samples representing 44 different types of mesenchymal tumors, Pacheco and Nielsen 10 observed that nuclear HDAC2 was intensively expressed in a very high proportion of sarcomas and was consistently high in translocation-associated sarcomas; for example, rhabdomyosarcoma (RMS), synovial sarcoma (SS), myxoid LPS, and endometrial stromal sarcoma (ESS), with an exception of low-grade fibromyxoid sarcoma. However, HDAC1 expression was low and weak in most sarcomas and was the lowest in translocation-associated sarcomas. 10 After normalization with control tissues, HDAC2 expression was consistently high in 77.5% of mesenchymal tumors, the highest in RMS, and the lowest in adamantinoma and chondrosarcoma (CHS). HDAC1 displayed heterogeneous expression, 37.5% with downregulation, 55% with upregulation, the highest in pleomorphic RMS and epithelioid sarcoma and the lowest in mesenchymal CHS, fibrosarcoma and well-differentiated LPS. There was no association between HDAC1 and HDAC2 expression (shown in Figure 1 and Supplemental data Table S1 and S2).
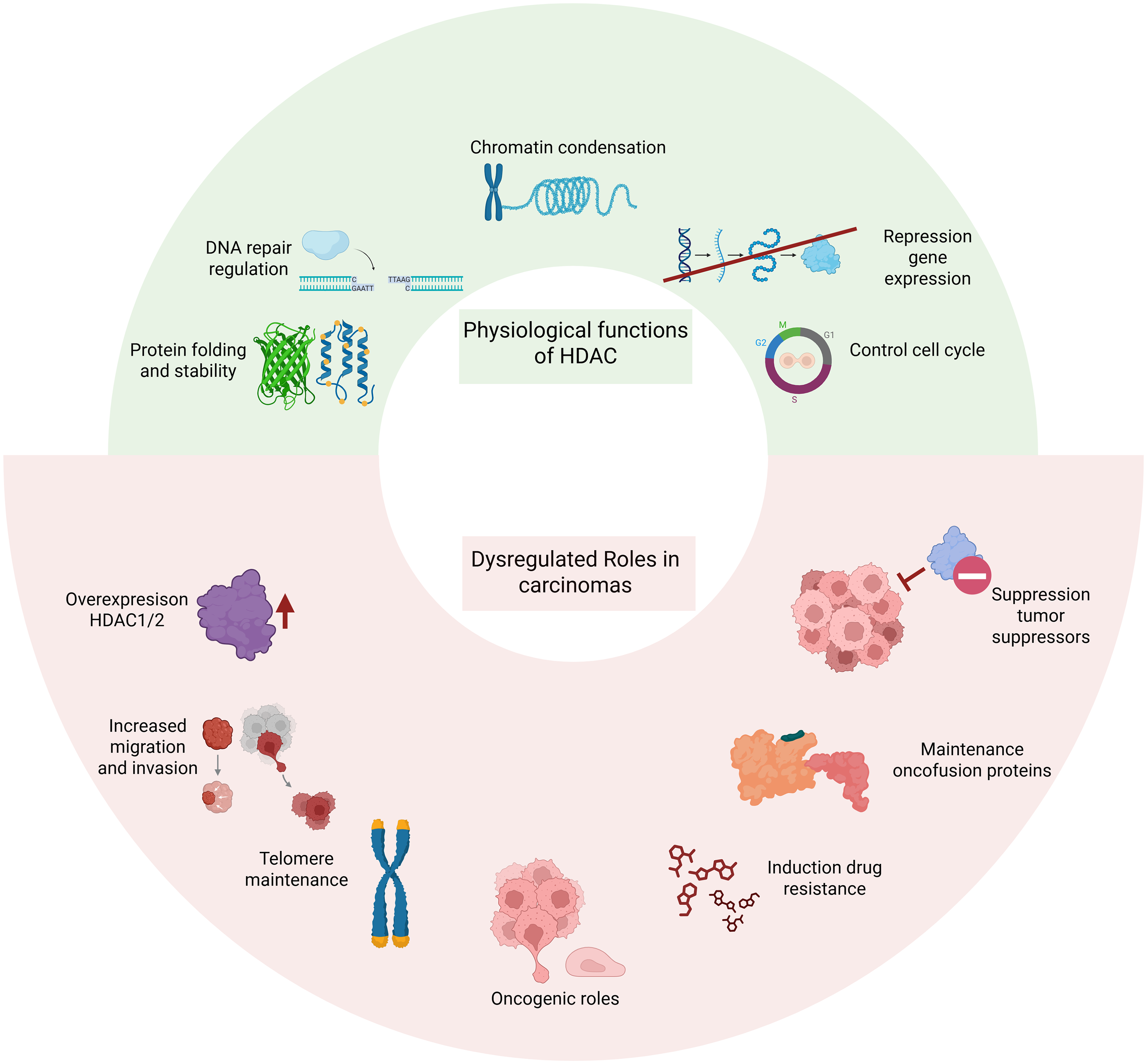
Physiological functions of HDAC and dysregulated roles in carcinomas. HDAC: histone deacetylase.
Changes in HDAC genes often co-occurred with the altered MAPK, PI3K, and ERBB pathways in sarcomas. Notably, HDAC alterations were found in 97% of 33 patients with the abnormal MAPK cascade, while in only 18% of 210 patients without the pathway alterations. 71 Deletion of either of HDAC1, 2, or 3—especially of HDAC1—strongly induced apoptosis of fibrosarcoma and LMS cells. Correspondingly, their gene expression levels were negatively correlated with overall survival (OS) of STS patients. 20
Alterations of HDAC expression and function in specific sarcoma types
Bone sarcoma
Osteosarcoma
Osteosarcoma, the most frequent primary malignancy of bone, appears mostly in children and adolescents. 3 All HDAC1–11 genes were expressed in osteosarcoma tissues. 41 Protein levels of Class I HDAC1–3 were elevated. Correspondingly, increased nuclear HDAC (but not HAT activity) caused decreased HAT/HDAC activity ratio in 49% of sarcoma tissues and 69.9% of cell lines. 13 HDAC2 was particularly high in sarcoma tissues and cells at mRNA and protein levels.11,13 Chaiyawat et al. reported the positive expression of nuclear HDAC1–3 proteins in 82%, 99%, and 93% of 89 patients, respectively. In 55% of the cases, HDAC2 (>8) was detected, whereas HDAC1 showed weak staining in 70%. 15 Another study even showed upregulated HDAC2 protein expression in 95% of 20 patients, mainly localized in nuclei and cytoplasmic perinuclear (both strongly stained >7). 16 However, downregulated nuclear HDAC2 protein was also detected in 20 patients. 10 Additionally, upregulated class II HDAC 4, 5, and 9 were found both at mRNA and protein levels,17–19 and HDAC4 was co-overexpressed with PCNA in osteosarcoma tissues. 17
An analysis of the TCGA database proved the HDAC2 gene expression level as a negative prognostic predictor for osteosarcoma. The patients with upregulated HDAC2 showed strongly reduced OS. 13 Conversely, Chaiyawat et al. reported that low levels of HDAC1–3 proteins indicated poor survival. Particularly, low levels of HDAC1 were associated with a high Enneking stage and the presence of initial metastasis. Five-year survival rates were 19% in patients with low HDAC1 level, but increased to 40% in patients with high HDAC1 levels. Low levels of HDAC3 were correlated with age >15 years. 15
HDAC 1 and 3 promote osteosarcoma cell growth and survival. HDAC1 knockdown caused osteosarcoma cell cycle arrest, loss of mitotic cells and caspase-3-dependent apoptosis, accompanied by upregulated apoptosis genes and altered cell proliferation genes. HDAC3 deletion reduced osteosarcoma cell growth and induced the greatest gene alterations (1317 genes), compared to 987 gene alterations due to HDAC1 ablation. In addition, only 31% of genes altered by HDAC3 were in common with those regulated by HDAC1, 39 indicating non-redundant roles of two HDACs in osteosarcoma.
HDAC1 is recruited by metastasis associated protein MTA1 to bind and deacetylate the stress response gene activating transcription factor ATF4, leading to inhibited ATF4 ubiquitination and increased ATF4 stability and activity in osteosarcoma. Upregulated ATF4 further forms a positive feedback loop with HDAC1 and MTA1 to enhance their expression and sarcoma growth. 38 HDAC2 increases p65 phosphorylation and nuclear accumulation, resulting in IL-6, MMP-2 and MMP-9 induction and osteosarcoma cell migration. 13 The HDAC1/2 heterodimer interacts with C-terminal-binding protein CtBP1 and interferon regulatory factor IRF1 to inhibit growth arrest specific 5-mediated signaling. The phenotype of osteosarcoma cells was reversed following either IRF1 overexpression or CtBP1 knockout. 40 Different from other class I HDACs, HDAC8 expression was even low in osteosarcoma. HDAC8 activation was found to inhibit the osteosarcoma cell proliferation via increasing p53 expression and inhibiting the STAT3/ERK activation. 43
Class IIa HDAC 4, 5, and 9, and class IIb HDAC6 also promote osteosarcoma malignancy. HDAC4 interacts with proliferating cell nuclear antigen (PCNA) and inhibits the PCNA ubiquitination. 17 Many microRNAs (e.g., miR-140,44,45,72 miR-145-3p, 73 and miR-591 46 ) downregulate HDAC4 expression to inhibit osteosarcoma growth. In contrast, the long noncoding RNA MALAT1 and circular RNA ABCC1 (circABCC1) upregulate HDAC4 expression via targeting microRNAs. MALAT1 regulated HDAC4 mediated proliferation and apoptosis via decoying of miR-140-5p. 44 Similarly, circABCC1 targets miR-591. 46
HDAC5 promotes sarcoma growth via inducing twist 1 expression. 18 HDAC5 maintains the length of long telomers. Upon depletion of HDAC5 shortened longer telomers and homogenization of telomere length were observed, predisposing osteosarcoma cells to chemotherapy. 47 HDAC9 binds to p53 proximal promoter region and deacetylates histone H3 at p53 loci, which suppresses the p53 transcription, thereby enhancing the osteosarcoma cell proliferation and invasion. 19
HDAC6 plays multiple roles in osteosarcoma biology. HDAC6 functions as a downstream effector of ROCK-TPPP1 and is inhibited by TPPP1. After ROCK activation, TPPP1 is phosphorylated, deactivating the inhibition. Activated HDAC6 deacetylates microtubule and β-catenin to enhance the cell migration and invasion, 49 and the expression of β-catenin and c-myc. 50 Moreover, through the activated STAT3 pathway, HDAC6 upregulates the expression of program death receptor ligand PD-L1, an important co-stimulatory molecule that activates the PD-1 inhibitory regulatory pathway in T-cells. 51 Additionally, HDAC6 contributes to chemoresistance to Adriamycin (ADR), doxorubicin, and cisplatin (CDDP). HDAC6 levels were significantly high in chemoresistance osteosarcoma cells.53,54 HDAC6 forms a complex with RUNX2 and p53 to suppress p53, p21, and BAX transcription, which causes reduced apoptosis in cells treated with ADR. 52 HDAC also upregulates IL-8 and ABCB1 transcription via p65 activation, 53 or binds with estrogen-related receptors alpha (ERRα) to regulate ERRα acetylation and increase its protein stability. 54
Alternatively, HDAC2 inhibits the osteosarcoma pluripotency. HDAC2 knockdown induced upregulation of stemness markers Sox2, OCT4, Nanog, and CD133, and DNA methylation. The osteosarcoma cells lacking HDAC2 formed more sarcospheres and colonies in vitro, grew faster and generated bigger tumors in vivo in nude mice. 16 However, this observation was argued due to HDAC1 compensation. 41 On the contrary, Senese et al. 39 found that HDAC2 knockdown had no significant effect on osteosarcoma cell U2OS growth, with a mild phenotype change and the fewest gene alterations (283 genes) related to cell proliferation and apoptosis, compared to HDAC 1 or 3 ablation. Moreover, HDAC1 silencing altered 987 genes, which was decreased to 608 genes by HDAC1/2 double deletion. Increased p21, and downregulated cdc25c, CCNG2, NF1A, SEP6 expression by HDAC1 knockdown were abolished by HDAC1/2 double silencing, 39 suggesting opposite roles of two HDACs in osteosarcoma.
Moreover, as a p53 co-activator, HDAC2 is co-localized and interacts with p53 and ATM in the nucleus, and participates in the early molecular events following DNA damage in osteosarcoma. Its silencing attenuated cell death, together with reduced p53 accumulation and activation in sarcoma cells treated with ADR. 42 Similarly, HDAC5 also plays dual roles in osteosarcoma. HDAC5 overexpression promoted the expression of apoptosis genes associated with the tumor necrosis factor receptor death pathway, combined with a decrease in proliferation genes within the MAPK pathway. 48 Notably, HDAC suppresses the Notch pathway in osteosarcoma. A higher abundance of notch pathway gene was found in potentially metastatic osteosarcoma cells. HDACi valproic acid (VPA) at therapeutic concentrations induced Notch genes expression causing a 250-fold increased invasiveness of non-invasive osteosarcoma cells. 73
CHS
CHS is the most common primary solid bone tumor and shows an incidence rate of about three cases per 1 million individuals annually. It contains 90% of conventional CHS and very rare subtypes: dedifferentiated, mesenchymal, clear cell, and extra-skeletal myxoid CHS. 74
Both nuclear HDAC 1 and 2 protein expressions were down in CHS, except upregulated HDAC2 in translocation-associated extra-skeletal myxoid CHS. HDAC1 was particularly low in extra-skeletal myxoid and mesenchymal CHS. 10 Venneker et al. 8 reported that HDAC1–11 isoforms were expressed in all seven tested CHS cell lines, wherein HDAC1–3 were the most abundant isoforms and indispensable for CHS cell growth. 8 Cytoplasmic HDAC6 was overexpressed, accompanied by strong Ki67 staining and low levels of cilia-related structure protein IFT88 in CHS tissues. IFT88 expression and the formation of the primary cilia are suppressed by HDAC6, leading to growth and invasion of CHS upon activation of Aurora A. 34
Unlike other HDACs, HDAC4 is a CHS suppressor. Reduced HDAC4 expression, but upregulation of Runx2 and vascular endothelial growth factor (VEGF) was found in CHS cells. HDAC4 binds to and deacetylates Runx2, leading to reduced Runx2 and VEGF expression. 35 Wnt3A/β-catenin pathway targets HDAC4, leading to HDAC reduction, but upregulated MMP3 and MMP13 expression. 36
EwS
EwS is a highly malignant bone and STS with early metastasis to lung and bone, accounting for 5% of all child and adolescent cancers. Almost all EwSs contain an ETS rearrangement, including EWS-FLII (90%) or EWS-ERG (5–10%) gene fusions. 75
Class I HDAC1–3 and 8 genes were constitutively expressed in EwS tissues and cell lines. 11 HDAC1 expression was low at mRNA 11 and protein levels, 10 while HDAC2 and 3 genes 11 and nuclear HDAC2 protein 8 were upregulated in EwS tissues. HDAC6 showed heterogeneous expression in the nucleus and the cytoplasm of EwS tissues. 12 The expression levels of individual class I HDAC gene 11 and HDAC6 protein 12 were negatively associated with event free survival and OS of EwS patients.
Class I HDACs promote cell proliferation, invasion, and local tumor growth, wherein HDAC 3 and 8 have the weakest roles. Deletion of either HDAC1 or 2 induced apoptosis and double-strand breaks, and profoundly reduced colony formation and EwS cell growth. However, cell growth was only slightly inhibited by HDAC3 knockdown and even less by HDAC8 knockdown. HDAC8 with HDAC1 synergistically promotes EwS. HDAC1/8 double deletion, though not of HDAC8 alone, strongly reduced EwS cell growth and invasion, superior to single HDAC1 deletion. 8
HDAC3 maintains EwS viability and genomic stability via the EWS-FLI1/HDAC3/HSP90 axis. EWS-FLI1 binds to and activates HDAC3 transcription. HDAC3 knockdown or inhibition induced HSP90 acetylation and reduction of its clients BRCA1, BRCA2, and RAD51 (major components of the homologous recombination DNA double-strand breaks repair pathways), leading to loss of genomic stability and SwS cell death. 37 Moreover, nuclear, but not cytoplasmic HDAC6 deacetylates Sp1, and enhances the SP1/P300 activator complex binding to EWSR1 and EWSR1-FLI1 promoters, thereby promoting EWSR1 and EWSR1-FLI1 transcription. Moreover, nuclear, but not cytoplasmic HDAC6 deacetylates Sp1, and enhances the SP1/P300 activator complex binding to EWSR1 and EWSR1-FLI1 promoters, thereby promoting EWSR1 and EWSR1-FLI1 transcription. HDAC6 inhibition impaired this binding, thereby inducing EWSR1-FLI1 downregulation and reducing in vitro and in vivo EwS growth. 12
STS
LMS
LMS is one of the most frequent STSs in adults, with an incidence ranging between 10%–20% of all newly diagnosed STSs. It can occur in almost any part of the body. 76
HDAC amplification was common in LMS. HDAC1–11 genes were overexpressed in 75.25% of 101 LMS patients (TGCA database). HDAC 3, 6, 10, and 11 were the most abundantly amplified isoforms, ranging between 19–23%. HDAC1 was amplified in 14% and HDAC2 in 9% of cases. 20 Accordingly, nuclear HDAC 1 and 2 proteins were highly expressed in 131 LMS tissues and HDAC2 expression was higher in LMS from soft tissues than from other locations. 10 The gene expression level of HDAC1 or 2 negatively predicated OS, and that of HDAC3 also showed a trend toward a shorter OS. 20 Class IIa HDAC 4, 5, and 9 overexpression was observed in 22–30% of 100 LMS patients. 23 HDAC9 was the most highly expressed and its truncated isoform MITR (without catalytic domain) were expressed in 92.3% of patents with high myocyte enhancer (MEF) 2 expression.22,23 HDAC4 protein expression was found in the majority of these patients, albeit 46% with weak and diffused/pan HDAC4 expression, 19% with an intense diffused signal; however, 8% had prominent nuclear accumulation. 21 The gene expression levels of HDAC 4 and 9, together with MEF2 were negatively associated with OS. Higher HDAC4 levels were in tumors featuring higher proliferative activity (Ki67 and mitotic index). A negative correlation between the HDAC9 and the Fas expression was found.22,23
Class II HDAC4, 7 and 9 play the crucial roles in LMS malignancies via regulating MEF2 target genes.21–23 They generally shuttle from cytoplasmic to nuclear and perform different functions. HDAC4 regulates gene expression involved in oxidative stress responses, proliferation, and apoptosis in LMS. 22 Cytoplasmic HDAC4, 5, and 7 regulate normal muscle cell differentiation at different stages, 77 while nuclear active HDAC 4 and 7 suppress MEF2 target gene expression and induce transformation of normal fibroblast cells. The transformed cells were characterized by increased proliferation potential and an overt cytoskeleton reorganization, which overcame the contact inhibition, grew in soft agar, and formed in vivo tumors in nude mice. 21 Additionally, HDAC4 contributes to senescence resistance of LMS cells. HDAC4 depletion induced senescence phenotype and the expression of a senescence gene set, leading to reduced LMS cell proliferation. 56
HDAC9 was co-overexpressed with MEF2D in LMS. HDAC9 forms a positive circuit with MEF2D which induces sustained cell proliferation and survival via suppressing the expression of FAS and some MEF2 target genes. HDAC9 is a regulator of cytoskeletal structure and fosters tumor cell motility. Silencing HDAC9 was connected to a decline in cell-peripheral F-actin accumulation, suppressed cell spreading, random migration, and the transformed phenotype of LMS cells.22,23
LPS
LPS, the most common human sarcoma, accounts for approximately 20% of STS, containing four principal subtypes: well-differentiated LPS/dedifferentiated LPS (WDLPS/DDLPS), myxoid/round cell and pleomorphic LPS (PLPS). 78
Altered HDACs expression was frequent in LPS. Somatic copy numbers of HDAC1–11 were amplified in 70% of LPS. HDAC 2 and 9 were the most frequently amplified, being 25% and 26%. HDAC1 was amplified in 16% and HDAC3 in 18% of cases. 57 Nuclear HDAC1 protein expression was low in most LPS (except PLPS), especially in WDLPS and myxoid LPS. 10 HDAC1 mutation also occurred in 8.3% of DDLPS. 25 On the contrary, nuclear HDAC2 protein expression was consistently high in low-differentiated and aggressive DD, myoxid and pleomorphic LPS, but not in WDLPS, 10 suggesting a prominent role of HDAC2 in LPS malignancy.
The gene expression levels of HDAC1–3 were negative prognosis for LPS patients.20,24 Patients with higher HDAC2 mRNA expression had markedly shorter disease-free survival (DFS), distant recurrence-free survival, and OS; for example, for median OS, HDAC2 high: 18.5 months versus HDAC2 low: 76.4 months. 24
As a downstream effector of EWSR1-DDIT3 (a unique fusion in myxoid LPS), HDAC1 selectively repress osteoblastic and chondrocytic transcription in multipotent mesenchymal cells. In the presence of EWSR1-DDIT3, HDAC1 is recruited to C/EBP sites within the promoters Opn and Col11a2, resulting in suppressed gene transcription. 57 HDAC2 was the most highly co-expressed class I HDAC isoform with MDM2. MDM2 promotes sarcoma cell survival via p53 degradation. Silencing or inhibition of HDAC2 reduced MDM2 expression, leading to p53 reactivation, apoptosis, and tumor growth inhibition. 24
Class II HDAC genes were found to be heterogeneously expressed in two independent LPS data sets (84 and 89 patients). HDAC4 gene expression levels were negatively correlated with MEF2 target genes expression in LPS in the presence of PTEN expression. 21
Rhabdomyosarcoma
Rhabdomyosarcoma (RMS), the most common pediatric STS arising from skeletal muscle precursors, is classified as fusion negative FN-RMS (including formerly embryonal RMS and pleomorphic RMS (PRMS), and more aggressive fusion positive FP-RMS (previously alveolar ARMS), due to the expression of PAX3- or PAX7-FOXO1 fusion proteins, preceded by chromosomal translocations.
Class I HDAC genes were constitutively expressed in RMS. 20 Nuclear HDAC 1 and 2 proteins were upregulated in all subtypes of RMS, and HDAC1 protein expression in PRMS was four times higher than in control tissues (shown in Figure 2). 10 In both FP- and FN-RMS tissues, nuclear HDAC3 protein was above 90% positively expressed 26 and cytoplasmic HDAC6 was in 94% positively expressed at variable intensity, 28 compared to their negative expression in normal skeletal muscle. HDAC5 mRNA level was also high in 137 primary FP-RMS. 27 RAC1 (a Rho family GTPase), a negative predictor of RMS, is co-localized with HDAC6, whereas HDAC6 level has no correlation with patient OS. 28
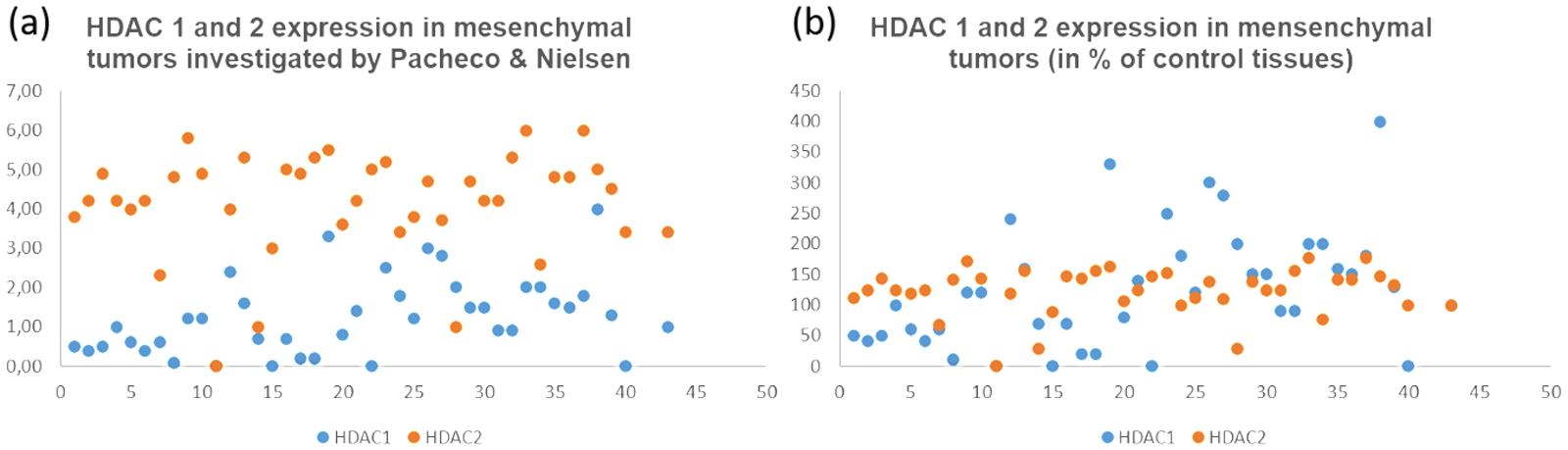
HDAC 1 and 2 expressions in mesenchymal tumors analyzed by Pacheco & Nielsen with IHC. (a) Mean scores of HDAC 1 and 2 immunostaining in 40 subtypes of mesenchymal tumors (including sarcomas and borderline tumors), and (b) after normalization with 22 normal mesenchymal tissues (vessels, muscle, nerve, and fat). X-axis in both: sarcoma types; Y-axis in (a): mean score of HDAC staining, and in (b): HDAC expression in percentage of control tissues. Detailed information of tumor subtypes was shown in table 1 1.
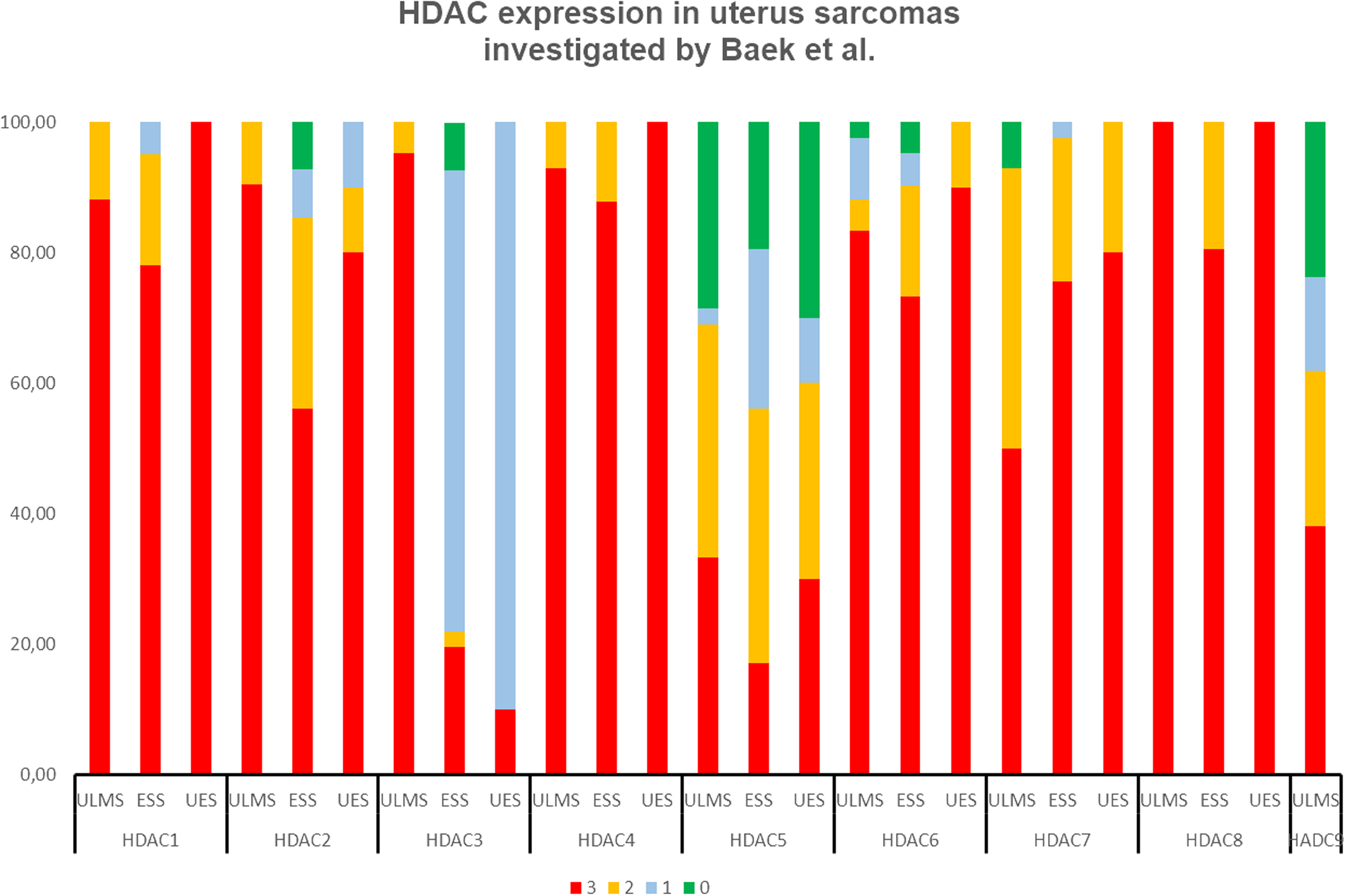
Prevalent HDAC1-9 overexpression in uterine sarcomas investigated with immunohistochemistry (IHC) by Baek et al, including ULMS (42 cases),2 ESS (41 cases)3 and UES (10 cases).4
All HDAC1–3 isoforms are essential for the integrity maintenance of core regulatory transcription factors (CR TFs).61,62 They interact with PAX3-FOXO1 and other CR TF (e.g., myogenic transcription factors MYOG, MYOD, and MYCN) at super enhancers (SEs). 79 SEs are the segments of DNA presenting the highest levels of both histone acetylation and HDACs, and oppositely regulate CR TFs. Hyperacetylation halts CR TFs transcription, while HDAC binding is positively correlated with CR TF networks. 61 All HDAC1–3 isoforms are indispensable for preventing the spreading of H3K27ac beyond SE boundaries and the following loss of prominent SE to SE interaction, and for promoting the binding of RNA Pol2 and CR TFs at SEs. Genetic depletion or inhibition of all three HDACs, but not single HDAC isoforms, completely inhibited CR TFs transcription.61,62
HDAC1–3 upregulate PAX3-FOXO1 protein abundance. Knockdown or inhibition of HDAC1–3 reduced PAX3-FOXO1 protein levels. HDAC3 particularly promotes PAX3-FOXO1 protein expression via upregulated chromatin remodeler SMARCA4 and downregulated miR-27a. 63 Additionally, HDAC3 suppresses RMS myogenic differentiation through the NCOR/HDAC3 transcriptional repressor complex. Knockout of HDAC1–3 reduced cell proliferation, while only HDAC3 deletion elevated expression of key myogenic regulatory genes and induced myogenic differentiation to varying degrees in a panel of FP- and FN-RMS cells. 26 HDAC1 also suppresses PTEN, p21, p14, and muscle-specific gene transcription due to the recruitments to their promoters by oncogene TBX2, leading to inhibited terminal differentiation and enhanced RMS cell growth.58,59 Additionally, HDAC1-2 forms a complex with SNAIL to suppress the MYF5 expression and myogenic differentiation of FP-RMS cells. 60
Besides HDAC3, HDAC4 knockout induced RMS differentiation and growth reduction. HDAC4 might function as scaffolding molecules to recruit HDAC3 to transcriptional target genes involved in myogenic differentiation in RMS. 26 HDAC 4 and 5 preferentially interacted with MEF2Cα1 (without myogenic activity), yet not with the muscle-specific MEF2Cα2. MEF2Cα1 recruits HDAC 4 and 5 to the promoters of muscle-specific genes MOD2, TNNI2 and p21, resulting in suppressed cell differentiation. 66 HDAC5 also binds to PAX3- and PAX7-FOXO1 to increase IL24 expression. 80 The binding sites were confirmed by gene analysis, 81 and PAX3 or PAX7-FOXO1 overexpression upregulated HDAC5 expression in FN-RMS cells. 27 Moreover, HDAC6 deletion strongly reduced RMS cell growth. 26 HDAC6 promotes growth, self-renewal, migration, and invasion of RMS cells via upregulating RAC1 transcription, which is co-localized with HDAC6 in cell membrane and folds. This facilitates cell cycle progression, enhances stem cell marker expression, inhibits cell differentiation, and promotes cytoplasmic actin reorganization. 28
Alternatively, HDAC suppresses oncogene ezrin expression. The ezrin gene promotor showed elevated acetyl-H3-K9 in the highly metastatic RMS cells. HDACi trichostatin A (TSA) increased ezrin expression and stimulated the lung metastasis of poorly metastatic RMS cells characterized by low ezrin levels, but not of high metastatic RMS cells with high ezrin expression. 82
SS
SS, a high-grade STS, accounts for 5–10% of all STS, occurring predominantly in older children and young adults, characterized by the fusion oncogene SS18:SSX, caused by the translocation t(X;18). 83
HDAC1–3 and 8 genes were constitutively expressed in SS tissues. 11 HDAC1-22 genes were the most abundantly expressed in SS cells 68 and their proteins levels were high in SS tissues. 10 HDAC2 expression was particularly high at mRNA and protein levels in SS cells and tissues.10,11,68
Both HDAC1 and 2 are involved in SS genesis driven by SS18-SSX fusion.67,84 HDAC1 contributes to SS18-SSX-mediated gene regulation via its association with TLE1 and EZH2. TLE1 recruits HDAC1/EZH2 complex to SS18-SSX target promoters, thereby inhibiting the tumor suppressor expression. Depletion or inhibition of HDAC1 reversed the epigenetic silencing of EGR1 and PTEN, induced apoptosis and reduced SS growth.66,85 However, HDAC1 also interacts with SS18-SSX, TCF/LEF and TLE to inhibit Wnt target gene AXIN2 expression. HDACi TSA upregulated AXIN2 expression. 67 HDAC2 acts as a safeguard to protect the SS18-SSX fusion from ubiquitin-mediated degradation. HDAC2 depletion strongly inhibited the interaction of MDM2 with HDAC2 downstream substrate, MCL-1 ubiquitin ligase E3 (MULE). It resulted in enhanced MULE protein stability and accumulation, leading to SS18-SSX degradation. 68
Uterine sarcomas
Uterine sarcomas account for approximately 3–7% of all uterine cancers, including four common subtypes: uterine carcinosarcomas (50%), uterine LMS (ULMS, 30%), ESS 15%, and undifferentiated endometrial sarcomas (UES, 5%). 86 ULMS has different tissue origin and molecular profiles from other soft tissue LMS (STLMS), sharing an overlap of only 210 genes with STLMS. 87
Deregulated HDACs in uterine sarcomas
Class I HDAC1–3 mRNA and protein are moderately expressed in normal endometrium throughout the menstrual cycle, while their expression is strong and mostly linked to high cell proliferation, high tumor grade, and short survival in several uterine malignancies, including uterine sarcomas.30,33,86,88
Upregulated HDAC2 protein expression was observed in 22 ESS and in 2 UES. Nuclear HDAC2 expression was positive in all sarcoma, being high in 36.3% of ESS and in 100% of UES, compared to 5% of normal endometrium. However, nuclear HDAC1 protein expression was weak and not significantly different from normal tissues. 33 This observation is in accordance with the observation of 32 ESS patients from Pacheco and Nielsen. 10 Baek et al. 30 thoroughly analyzed the expression of HDAC1–9 proteins in uterine sarcomas (an overview shown in Figure 2). The strong expressions of HDAC1–4, 6 and 8 proteins were prevalent in 42 ULMS tissues, ranging between 83.3–100%, while the strong expression of HDAC 5, 9, and 7 proteins was infrequent, ranging between 33% and 50%. 30 In agreement with this, another research demonstrated an 8.4% gain of HDAC9 in 15 ULMS by array-CGH analysis. 1 In ESS and UES, most HDACs proteins were highly expressed in a similar pattern, though higher in UES than in ESS. Upregulated HDAC 1, 4, and 6–8 expression ranged between 73.2% and 87.8% in ESS, and between 80% and 100% in UES. An increase in HDAC2 expression was reported in 56.1% of ESS, and in 80% of UES. In contrast, high levels of HDAC 3 and 5 were infrequent. The strong staining was detected in <30% of ESS and UES patients.31,32
Clinical relevance and roles of HDACs in uterine sarcomas
Low levels of HDAC1–9 (especially class IIa HDAC 5, 7, and 9) were potentially good prognostic markers in ULMS, in the presence of p53 expression or non-epithelial subtype. The low level of cytoplasmic HDAC5 in combination with epithelioid subtype was an independent predictor for DFS, identified by multivariate analysis. 30 Di Giorgio et al. 23 found that both HDAC 4 and 9 promotes ULMS, while HDAC9 particularly contributes to malignancies of ULMS cell Sk-UT-1 via binding to the promoters of some MEF2s target genes to suppress their expression. HDAC4 specifically suppresses ALPK2 and IL8 expression, a non-redundant role different from HDAC9 in ULMS. 23
In ESS, HDAC expression had no association with pattern of recurrence, lymph node metastasis, tumor size, and FIGO stage, while a high frequency of HDAC 1, 4, and 6–8 strong expression showed a trend associated with a high rate of recurrence and a low 10-year DFS. 32 In UES, the strong expression of cytoplasmic HDAC6 and CYP19A1 were associated with distant recurrence. 31
HDAC2 plays an essential role in enhancing proliferation and suppressing differentiation, yet not in maintaining survival of ESS cells. HDAC2 reduction by VPA increased p21 expression, leading to growth inhibition and differentiation, though not to apoptosis in ESS cells. 33 Akt activation was inhibited by HDACi SAHA and resulted in an induced mitosis failure with a subsequent autophagic ESS and apoptotic UES cell death. The effect was caused by a decrease of HDAC7 in ESS and a decline in HDAC 2, 3, and 7 in UES,69,70,89 suggesting that HDAC7 alone or together with HDAC2 and HDAC3 might maintain uterine sarcoma cell survival. The report is congruent with HDAC7 over-expression linked to ESS recurrence. 32
Cytoplasmic HDAC8 is a diagnostic marker for uterine tumors with smooth muscle differentiation. HDAC8 was highly localized in the cytoplasm in ULMS, leiomyoma, and some endometrial stromal tumors with smooth muscle differentiation, with a similar expression frequency to all smooth muscle markers.90,91 Cytoplasmic HDAC8 was strongly expressed in uterine sarcomas30–32 and had a trend of being associated with ESS recurrence, 32 while its role in uterine sarcoma pathogenesis remains unclear.
Conclusion and clinical significance
The frequent HDACs overexpression and their crucial roles in sarcomagenesis indicate the potential clinical application of HDACi. Consistently, HDACi emerged from several high-throughput drug screens, being active across different sarcoma types.9,62,92 HDACi panobinostat, romidepsin, quisinostat, and entinostat were identified as efficient drugs for osteosarcoma. 92 LPS, 93 RMS, 62 and quisinostat were found to be the most potent for SS. 94 The anti-tumor efficiency of HDACi was also demonstrated in numerous preclinical investigations with various sarcoma cells.95–97 However, the therapeutic benefits of HDACi (as monotherapy or in combination therapy) were modest in clinical trials. Generally, due to the rarity of sarcoma, patients with various sarcomas without stratification were enrolled in the trial using one HDACi, mostly one of class I, or pan-HDACi. For example, 40 patients with 20 different sarcomas in a phase II clinical trial (NCT00112463) were treated with HDAC1-2i romidepsin. The outcome showed that only one patient completed the trial, and 30 patients stopped due to progression.
Several factors are important for patients’ stratification. First, the expressions levels of HDACs, especially those of HDAC1–3, are plausible pre-selection biomarkers to identify the responsive tumor types. 98 The expression level of HDAC1 in LPS cells was associated with sensitivity of pan HDACi quisinostat. 93 The STS patients with high expression levels of class I HDAC showed better responses to class I HDACi chidamide. 20 However, HDACs overexpression was not prevalent in sarcomas, except uterine sarcomas. In maximally 30% of STS 20 and in up to 3% of bone and STS, 71 HDAC1–3 overexpression was found. Second, the expression profile and roles of HDACs vary in sarcomas (summary shown in Table S3). Most sarcomas had HDAC1 and 2 overexpression, while CHS only had HDAC6 upregulation, and downregulation of HDAC1 and 4 (summary shown in Table S3 and S4). Correspondingly, HDACs play cell context dependent roles. HDAC4 suppresses CHS but promotes other sarcomas. HDAC8 inhibits osteosarcoma cell proliferation, 43 but enhances EwS cell growth. 11 HDAC2 and 5 even play dual roles in osteosarcoma. Therefore, non-specific inhibition could mitigate, neutralize, or even reverse HDACi efficiency. Gene alterations due to HDAC1 deletion were evened out when combined with HDAC2 deletion in osteosarcoma. 39 HDACi VPA and TSA increased the metastasis potential of non-invasive RMS and osteosarcoma cells.73,82 Panobinostat combined with proteasome inhibitor bortezomib induced in vivo fast growth of orthotopic patient-derived RMS xenografts. 9 HDACi increased oncofusion EWS-FlI1 transcription activity and protein stability, and enhanced its binding to DNA.95,99 Third, sarcomas have different sensitivity to a certain HDACi. EwS was the most sensitive to HDAC6i BML-281. 12 SS cell SYO-1 was 300 times more sensitive to romidepsin (FK228) versus osteosarcoma cell SAOS2. 100 The clear cell sarcoma and EwS cells, both harboring the EWSR1 gene as a fusion gene partner (EWSR1::ATF1 and EWSR1::FLI1), exhibited higher sensitivity to vorinostat, compared with several epithelioid sarcoma, fibrosarcoma, SS, and LPS cell lines. 101
An accurate detection of the HDAC expression is the prerequisites for the efficient stratification of patient. Most HDACs are expressed to a varying degree in a multitude of normal human tissues including stromal and inflammatory cells 102 ; therefore, IHC is the easiest and the most accurate way to analyze cell localization and tissue distribution of HDAC. 98 However, using IHC analysis, there still exist some discrepancies among different studies,32,33 which might be due to several reasons. First, due to heterogeneity of tumor tissues, HDACs expression levels could vary considerably between tumors of the same entity. 102 Second is the specificity of the antibody. A strict standard for the HDAC antibody selection was Western blot analysis, being used in several studies.10,33,70 Third is the quantitation of HDACs expression in different location; for example, in the nucleus, in the cytoplasm, or both.
Besides HDAC, other molecular markers could predict HDACi response. GYS1 (involved in glycogen metabolism) is a biomarker for good response. Pathways involved in DNA replication, mitosis, and cell cycle are biomarkers for poor response of LPS cells to JNJ-26481585. Highly proliferative cells were more resistant to JNJ-26481585. 93 In sarcoma, SAHA sensitivity was associated with HR23b protein levels. Elevated HR23b expression was detected in only 12.5% of sarcomas—including malignant peripheral nerve sheath tumors, angiosarcoma, dedifferentiated liposarcomas, synovial sarcomas, and leiomyosarcomas—and in 23.2% of gastrointestinal stromal tumors. 103 A two gene set signature (high CUGBP2; low RHOJ) in sarcoma was associated with the synergistic combination efficiency of HDACi with DNA-methyltransferase inhibitors in vitro and in vivo. 104
Other factors are needed to consider. The preclinical study with mouse sarcoma cells might not be directly translated into clinical trials because of different HDAC expression profiles. Mouse osteosarcoma cell K7M2 exhibited higher levels of HDAC1–3 mRNA, but lower levels of HDAC 5 and 6 mRNA, compared to human osteosarcoma cell SAOS2. Pan HDACi or HDAC1-2i efficiently inhibited growth and lung metastasis, while silencing of either HDAC5 or 6 significantly increased in vitro growth of K7M2.
104
Moreover, the contribution of HDACs to tumorigenesis and tumor progression may not necessarily be related to its expression level, because aberrant HDAC activity is also common in the tumor development5,105; for example, increased HDAC activity in osteosarcoma.
13
Furthermore, the current HDACi might lack the potency to suppress sarcoma growth to a comparable extent to HDAC knockdown. HDAC3 deletion induced > 90% growth reduction and from 60% to 80% of RMS cells differentiation, while pan HDACi SAHA, TSA, or HDAC3i RGFP966 only induced a modest growth suppression and approximately 10
In summary, frequent amplification and/or overexpression of HDACs in sarcomas indicate that HDACi is a promising therapeutic option for sarcomas. A partial response has been shown in one ULMS patient treated with VPA combined with the VEGF inhibitor, bevacizumab, 106 which is particularly meaningful for uterine sarcomas because of prevalent HDAC expression. However, the different “double-edged sword” roles of HDAC indicate that in addition to HDACi, HDAC activation might be another direction for sarcoma therapy. Recently, HDAC8 activator TM-2-51 was found to suppress osteosarcoma progression in vitro and in vivo xenograft model. 43 Therefore, more studies on the expression pattern and role of the different HDACs isoforms in sarcomas are needed. 95 In summary, the diverse expression and roles of HDACs in sarcoma pathogenesis will be a solid baseline to guide the personalized therapeutic application of HDAC modulators in sarcoma patients.
Supplemental Material
sj-doc-1-jbm-10.1177_03936155251385254 - Supplemental material for Histone deacetylase in human sarcomas
Supplemental material, sj-doc-1-jbm-10.1177_03936155251385254 for Histone deacetylase in human sarcomas by Ping Quan, Christoph Schatz and Johannes Haybaeck in The International Journal of Biological Markers
Supplemental Material
sj-docx-2-jbm-10.1177_03936155251385254 - Supplemental material for Histone deacetylase in human sarcomas
Supplemental material, sj-docx-2-jbm-10.1177_03936155251385254 for Histone deacetylase in human sarcomas by Ping Quan, Christoph Schatz and Johannes Haybaeck in The International Journal of Biological Markers
Footnotes
Abbreviations
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.