
Abstract
Background
It is becoming increasingly evident that the vasculature is implicated in migraine pathophysiology. Calcitonin gene-related peptide (CGRP) acts as one of the key migraine mediators through various mechanisms that includes endothelium-mediated cerebral vessel vasodilation. Endothelial cells express mechanosensitive Piezo1 channels and have been suggested to play a role in migraine pathophysiology. However, the crosstalk between these two migraine-related signalling pathways remains unclear.
Methods
We measured intracellular calcium (Ca2+) in human induced pluripotent stem cell-derived endothelial cells (hiPSC-ECs), after exposure to Yoda1, a specific Piezo1 channel agonist, with and without CGRP. In addition, we investigated the effects of CGRP and Yoda1 on cellular remodelling by staining for focal adhesion (FA) protein paxillin using immunocytochemistry.
Results
Our data suggest that a one-hour sensitization of hiPSC-ECs with CGRP followed by application of Yoda1 leads to a higher intracellular Ca2+ level compared to when Yoda1 and CGRP are acutely applied separately or combined, suggesting at least indirect crosstalk between the two signalling pathways in the vascular system. CGRP receptor antagonist BIBN4096 significantly reduced the intracellular Ca²+ level under this sensitization protocol, confirming effective CGRP pathway blockade. The results also show that a one-hour sensitization of CGRP and Piezo1 activation affects cellular remodelling as evidenced by an increased number and area size of paxillin FA points per cell in hiPSC-ECs.
Conclusions
We have generated a human cell assay based on iPSC-derived endothelial cells and provided some evidence for crosstalk between mechanosensitive Piezo1 channels and CGRP in our hiPSC-EC system, which shows the potential for in vitro modelling of vascular implications relevant to migraine.
Introduction
Migraine is a neurovascular disorder characterized by recurrent disabling attacks of severe headache with associated features, such as nausea, vomiting, and enhanced sensitivity to sound and light (1). One-third of patients experience aura symptoms during an attack, which is why migraine is classified as either with aura or without aura (2). Despite recent advances in treatment options of migraine, which include antagonism of calcitonin gene-related peptide (CGRP) action, the treatment of many migraine patients remains suboptimal; hence it is important to better understand migraine disease mechanisms so that further improvements in therapeutic strategies can be made (3).
To date, migraine research is almost exclusively focused on experimental animal migraine models and collecting data from patient's studies. While these approaches have provided valuable insights, animal models are limited in their ability to fully replicate the human pathophysiology and human data typically lack depth with respect to mechanistic insight. This underscores the need for advanced experimental models. Human induced pluripotent stem cells (hiPSCs) offer new possibilities for brain disease modelling, enabling differentiation into various cell types for disease-relevant assays and drug screening (4–6).
The main difficulty in generating a meaningful hiPSC-based model for migraine lies in capturing the intricate interactions between vascular and neuronal components, both in the brain and at the level of the meninges. Current iPSC technology does not allow the generation of specific functional vessels such those in the meningeal vasculature, the sensory neurons that innervate them or the complex 3D multicellular system that is required to effectively study their interaction. In the present study, we used the main advantage of hiPSCs, which allow the dissection of a complex physiological vessel system with multiple various cell types and interactions with nerve endings to investigate a single component of that system, comprising the effects on human iPSC-derived endothelial cells (hiPSC-ECs). Such a reductionist approach enables the separation of individual roles of each cell type and their contribution to multiple migraine mechanisms before they are investigated in more complex in vitro or in vivo animal model systems or humans. One such approach is looking at the interaction between specific migraine-relevant signalling pathways relevant in hiPSC-ECs, with respect to model aspects of the brain vasculature, which is known as an important contributor to migraine pathophysiology.
Recent evidence suggests an interesting interplay within the neurovascular system between CGRP and Piezo1, through endothelial cells (7). CGRP is a very potent vasodilator and long known to be a primary mediator of migraine attacks (8). Its effect is likely mediated by binding to the CGRP receptor complex that is composed of calcitonin receptor-like receptor (CLR) and receptor activity modifying protein 1 (RAMP1) subunits, which are both expressed in endothelial cells (9). Activation of the complex by CGRP leads to intracellular cyclic adenosine monophosphate (cAMP) accumulation that triggers the release of diffusible messenger nitric oxide (NO), acting on the smooth muscle cells of the vessel, culminating in vasodilation (10). At the level of the meningeal vessels, this could contribute to neuronal sensitization (7), because NO donor glyceryl trinitrate is among the most effective inducer of migraine attacks in patients (11–13).
Endothelial cells also express Ca2+-permeable mechanosensitive Piezo1 channels, which, upon reacting to shear stress or blood vessel pulsation, can provide another input for Ca2+-dependent activation of nitric oxide synthase (NOS) and subsequent NO release (14). Based on this, Piezo1 channels were recently suggested to be a driver of migraine-related nociception, possibly explaining the pulsatile nature of headache and mechanical hyperalgesia (7,15,16). It should be noted that, during a migraine attack, CGRP-induced vasodilation leads to a closer proximity of cerebral vessels to nearby neurons, resulting in the mechanical activation of endothelial (and neuronal) Piezo1 channels (7). However, the pathways in endothelial cells are not yet explored in the context of migraine.
Apart from the Gas/cAMP/protein kinase A (PKA) pathway leading to NO production, CGRP is also known to activate phospholipase C (PLC), resulting in an increase in intracellular Ca2+ level in HEK293 cells. It has been suggested that the PLC pathway would provide an alternative mechanism of CGRP-mediated activation of NOS, independent of cAMP accumulation (10). This suggests some type of crosstalk of CGRP and Piezo1 pathways through intracellular Ca2+ increase needed for NO production to promote vasodilation (Figure 1). In addition, Zhang et al. 17 recently showed a dramatic increase in the activity of Piezo1 channels in endothelial cells induced by activation of the cAMP pathway, which is a classical signalling route also stimulated by CGRP. More specifically, they showed that endothelial Piezo1 channels are regulated by PKA and protein kinase C (PKC) phosphorylation at S1612, conserved phosphorylation site in Piezo1.
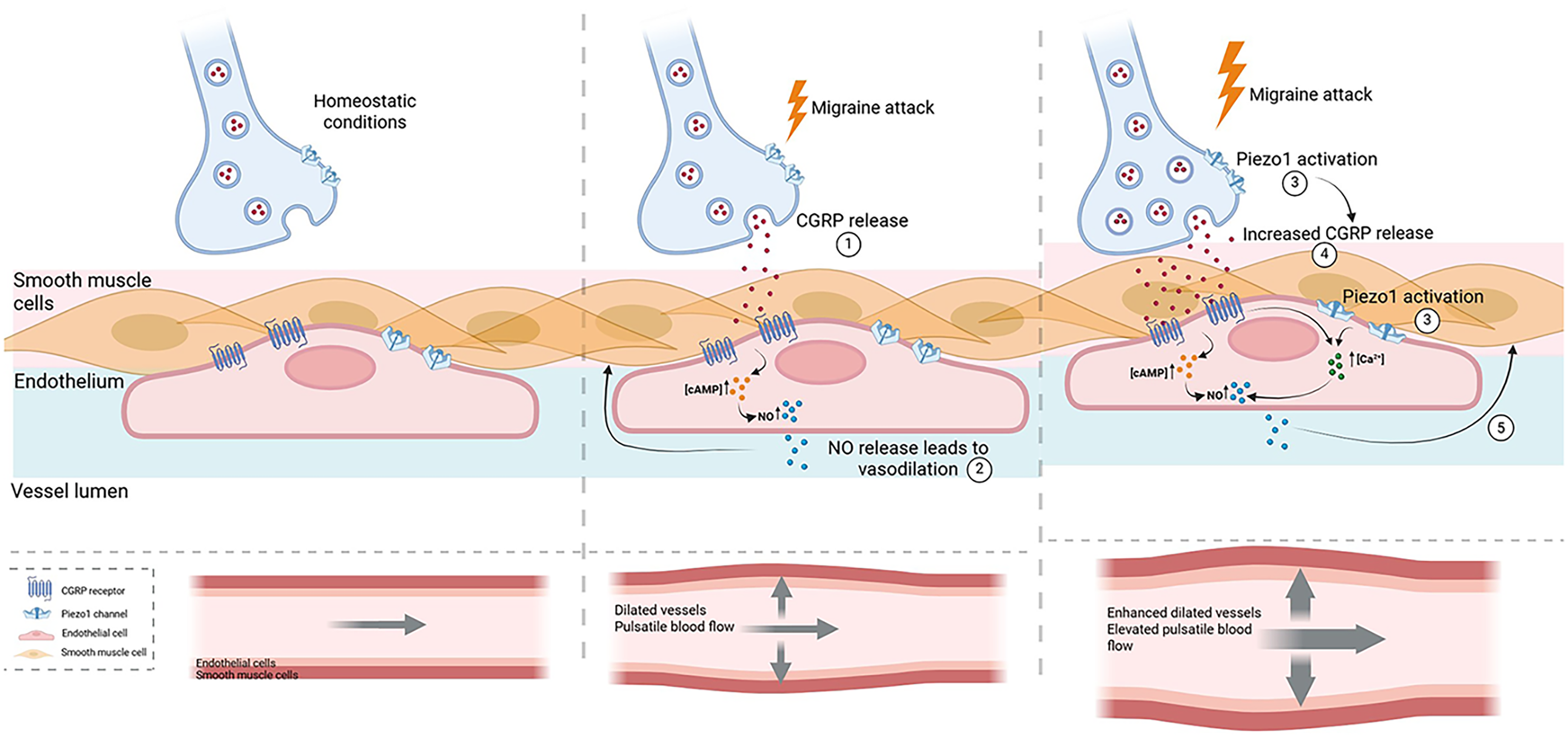
Schematic representation of the key elements in a migraine attack. During a migraine attack calcitonin gene-related peptide (CGRP) is released by neurons resulting in cyclic adenosine monophosphate (cAMP) accumulation and nitric oxide (NO) production in endothelial cells and subsequent vasodilation. During CGRP-induced dilation of the vessels, the extracellular space is reduced, and vessels come in closer contact with nearby neurons leading to the mechanical activation of neuronal and endothelial Piezo1 channels. Activated neuronal Piezo1 channels result in an increased CGRP release by neurons while endothelial activation of Piezo1 channels leads to an increased level of intracellular Ca2+. During this phenomenon, CGRP endothelial downstream pathways can also promote the increase of intracellular Ca2+ resulting in higher NO production. Higher NO release from endothelial cells leads to enhanced vasodilation and elevated pulsatile blood flow during a migraine attack. Figure created with BioRender.com.
Given the activation of the two signalling pathways, we hypothesize that cellular remodelling would take place by CGRP action (as seen with vasodilation) shown by reorganization of focal adhesion molecules (FAs), which in turn could lead to changes in cellular adhesion and growth through Ca2+ signalling (18). Paxillin is a key adaptor protein involved in the formation of FAs after being phosphorylated (19). It has been shown that, upon an increase in intracellular Ca2+ level or Piezo1 activation, paxillin's activation is affected, indicating the reorganization of FAs in endothelial cells (and neuronal cells) (20,21). Moreover, increased accumulation of paxillin at FA sites was shown to increase their size, and thereby enhance cell adhesion efficiency (22). Concurrently, the extracellular matrix (ECM) may stiffen, exhibiting increased resistance, in response to enhanced cellular forces aimed at optimizing adhesion, potentially due to local fiber alignment induced by mechanical stress (23).
The present study aims to elucidate interactions between CGRP- and Piezo1-mediated signalling in hiPSC-ECs to understand their possible role in migraine-related vasodilation. To this end, we explored effects of hiPSC-ECs stimulation by mechanosensitive Piezo1 channel agonist Yoda1 alone or together with vasoactive neuropeptide CGRP, with or without a CGRP antagonist. By simulating CGRP release and activating Piezo1 channels, we show the contribution of hiPSC-ECs to downstream intracellular pathways that ultimately lead to vasodilation through an increased intracellular Ca2+ level alongside Piezo1 mechanosensing channel activation. The increase of FAs of paxillin further suggest that CGRP and Piezo1 actions can locally affect reorganization of FAs and thereby cellular remodelling, which would lead to changes in the vasculature, such as vessel stiffness. We propose that this mechanism may have relevance to the aggravating pain sensation during pulsatile flow when CGRP is released locally and Piezo1 channels become activated.
Methods
hiPSC-ECs culture
As the hiPSC line, we used LUMC0020iCTRL06 (generated from skin fibroblasts; https://hpscreg.eu/cell-line/LUMCi026-A) (24). Research on hiPSC was approved by the medical ethical committee at Leiden University Medical Center (Leiden, The Netherlands). hiPSC-ECs were generated and maintained as previously described (25) with the slight modification of using a high concentration of 8 µ
Live Fluo-4 Ca2+ imaging
In case of sensitization, hiPSC-ECs were incubated with CGRP (1
Ca2+ imaging analysis
The Ca2+ signal was quantified using ImageJ 1.54d (National Institute of Health, Bethesda, MD, USA). Intensity values range from 0 to 255 as defined by the imaging software. For consistency, a fixed threshold value of 35 was applied across all videos using the colour threshold plugin with the intermodes filter to segment calcium signals. The Time Series Analyzer plugin was used to measure the average Ca2+ fluorescent intensity (FI) of 10 single cells per sample (20× magnification). Responders were identified based on peaks above baseline FI (ΔF > 2000 FI units). Fmax/F0 was analysed by dividing the maximum FI of each cell by its baseline average FI of the unstimulated recording, referred here as F0. Further analysis involved the time to reach Fmax, which was calculated based on the time recording of the videos.
Immunostaining
Cells were fixed with 4% paraformaldehyde for 10 minutes, permeabilized with 0.5% Triton-X100 for 10 minutes and blocked with 1% bovine serum albumin for 15 minutes. They were then incubated overnight at 4 °C with anti-paxillin (SAB4502553; Merck KGaA), anti-CD31 (PA5-32321; Thermo Fisher Scientific), anti-ZO1 (PA5-28858; Thermo Fisher Scientific), anti-VE-Cadherin-A488 (53-1449; eBioscience, Vienna, Austria) and anti-Piezo1 (NBP2-75617; Novus Biologicals, Abingdon, UK) antibodies (Table 1). After this, the cells were incubated with Texas Red anti-rabbit or anti-mouse immunoglobulin G for 1.5 h in the dark at RT (Table 1). The nuclei (NucBlue Live ReadyProbes, R37605; Thermo Fisher Scientific) and actin (ActinGreen 488 ReadyProbes, R37110; Thermo Fisher Scientific) were stained for 30 minutes in the dark at RT. Images were obtained using the Keyence BZ-X800 microscope.
List of antibodies and concentrations used in the present study.

Analysis of paxillin focal adhesion points
Paxillin images were randomly selected from six conditions: (i) Control, (ii) dimethyl sulfoxide (DMSO), (iii) Yoda1, (iv) CGRP, (v) CGRP + Yoda1 and (vi) CGRP 1h_Yoda1. For each condition, six images were analysed, approximately 200 cells per condition, with each image representing a distinct field of view. For the image analysis, we were blinded to the conditions. An automated pipeline was developed for localizing paxillin and quantifying its density in the confocal microscopy images. Initially, cell nuclei were segmented by applying intensity thresholding to blue channels with a threshold value of 100, where pixel intensities range between 0 and 255. Next, a connected component (CC) analysis was performed to discard CCs with small areas; the threshold was 70 pixels. This segmentation was used to mask out nuclei from paxillin images (red channel) for accurate localization.
For automated paxillin localization, we applied preliminary foreground segmentation followed by filtering out false positives. The preliminary segmentation of paxillin was carried out using the Chan-Vese active contours model (26) (MATLAB function “activecontour”). The Chan-Vese model was initialized with the regional maxima of the paxillin images with eight pixels connectivity (27). The speed function for the active contours was set to the gradient of the paxillin channel, with a contraction bias of −0.2. To prevent over-expansion, the maximum number of iterations was set to 5, ensuring the contours remained close to the locations of maxima. In Chan-Vese model, these initial contours deform on the speed function to adapt to the shape of the paxillin (28). The segmentation, however, can include false positives, which we removed by applying two additional filters: (i) CCs with an average intensity below 50 and (ii) CCs with areas larger than 10 pixels, as they were likely non-paxillin elements. An expert determined these thresholds by manually evaluating a few automated segmentation results. Once established, the thresholds were consistently applied across all images to ensure reproducibility.
For analysing the area size of paxillin FA points, the images showing only the red channel were converted to binary, to separate the paxillin stained areas from the background, using ImageJ 1.54d. After this, the “Analyze particles” function was applied with specific thresholds of area size between 0–6, 6–12 and 12–15 µm2. The scale of the software was fixed based on the scale bar of one sample image to acquire the precise measurements of area size (µm2) and it was globally applied to all the rest of the images included in the analysis. To avoid false positive selection of paxillin FA points, the selected regions of paxillin were manually filtered by cross-referencing the binary images with the corresponded raw images.
Statistical analysis
Statistical analysis was performed using Prism 6.01 (GraphPad Software Inc., San Diego, CA, USA). Data were analysed using an unpaired Student's t-test or one-way analysis of variance with Tukey's multiple comparisons test where appropriate. P < 0.05 was considered statistically significant.
Results
Characterization of hiPSC-ECs
HiPSC-ECs were cultured for three days in EC-CGM until confluency (Figure 2(A)). Immunostaining with endothelial markers showed that the cells exhibited the typical endothelial morphology with junctional localization of CD31, ZO-1 and VE-cadherin (Figure 2(B) to (D)). To confirm expression of Piezo1 channels, the cells were stained with anti-Piezo1 antibody. Figure 2(E) and (F) show the expression and localization of Piezo1 in the cells under normal culture conditions (white arrows). Piezo1 channels appeared to be present intracellularly, with higher expression around the nuclei in some cells (yellow arrows).

Characterization of hiPSC-ECs. (A) Phase-contrast image of the cells after three days of culture. (B–D) Representative images of CD31 (B), ZO-1 (C) and VE-cadherin (D) immunocytochemical staining (green or red) and 4′,6-diamidino-2-phenylindole (DAPI) (blue). (E) Representative image of Piezo1 immunocytochemical staining (in red) and DAPI (in blue) in hiPSC-ECs. (F) Zoomed-in area of interest (E). Scale bars, 200 µm (A), 100 µm (B-E), 50 μm (F).
CGRP exposure for 1 hour sensitizes Piezo1 channels leading to a higher intracellular Ca2+ level in hiPSC-ECs
To assess the triggering of the intracellular Ca2+ level after Piezo1 activation and CGRP binding, we performed live Ca2+ imaging. First, we conducted a concentration-response assay (0.01 µM

Application of Yoda1 and/or CGRP to induce intracellular Ca2+ levels in hiPSC-ECs. (A) Percentage of cells responding to Yoda1, CGRP, CGRP + Yoda1 and CGRP 1h_Yoda1 compared to controls (dimethyl sulfoxide (DMSO) vehicle control for Yoda1 and medium for CGRP). (B) Average maximum Ca2+ FI normalized by the average baseline Ca2+ signal (Fmax/F0). (C) Average time to reach maximum calcium intensity (Fmax). In (B) and (C), each dot represents one single cell. Yoda1: n = 6, CGRP: n = 6, CGRP + Yoda1: n = 6 and CGRP 1h_Yoda1: n = 6 (n = number of wells analysed). All results are presented as the mean ± standard error of the mean (SEM). *p < 0.05, **p < 0.01, ****p < 0.0001. Statistics performed by one-way analysis of variance with Tukey's multiple comparisons test.
BIBN4096 inhibits the action of CGRP leading to a reduction of intracellular Ca2+ level in hiPSC-ECs
To further confirm that CGRP indeed affects the intracellular Ca²+ level of the cells, we used BIBN4096, a reversible CGRP receptor antagonist. To this end, cells were pre-incubated with BIBN4096 for 30 minutes prior to CGRP treatment, and the antagonist was maintained throughout the experiment to ensure continuous receptor blocking. To confirm CGRP effects on intracellular Ca²+ levels and optimize antagonist concentration, we first performed a titration experiment testing a range of BIBN4096 doses (100 nM
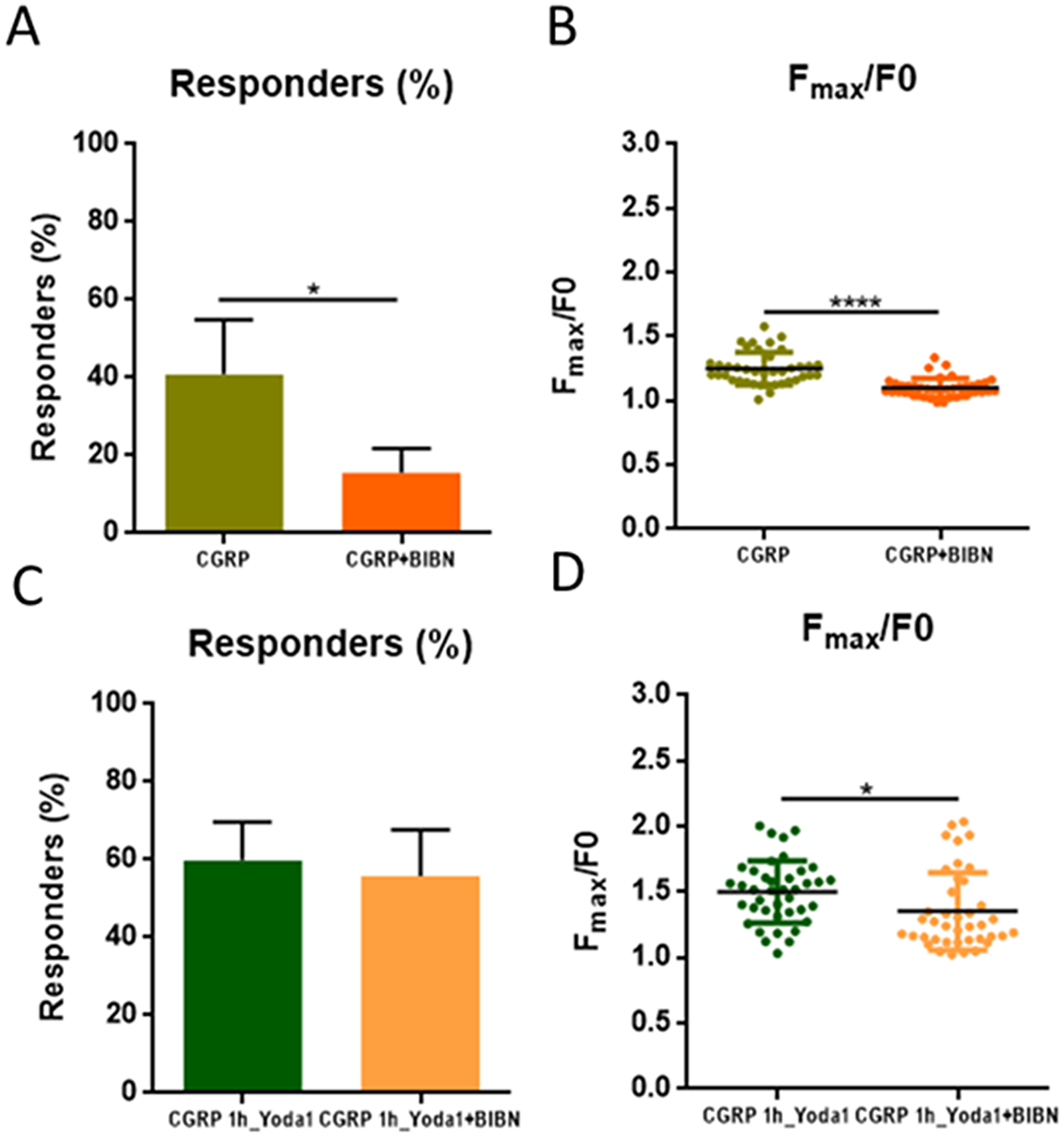
Application of BIBN4096 to reduce the intracellular Ca2+ levels of CGRP in hiPSC-ECs. (A) Percentage of cells responding to CGRP and CGRP + BIBN. (B) Average maximum Ca2+ FI normalized by the average baseline Ca2+ signal (Fmax/F0). (C) Percentage of cells responding to CGRP_1h_Yoda1 and CGRP_1h_Yoda1 + BIBN. (D) Average maximum Ca2+ FI normalized by the average baseline Ca2+ signal (Fmax/F0) in CGRP_1h_Yoda1 and CGRP_1h_Yoda1 + BIBN. In (B) and (D), each dot represents one single cell. CGRP: n = 4, CGRP + BIBN: n = 4, CGRP 1h_Yoda1: n = 4 and CGRP 1h_Yoda1 + BIBN: n = 4 (n = number of wells analysed). For all experiments involving BIBN4096, cells were pre-incubated with the antagonist for 30 minutes prior to treatment and BIBN4096 was maintained throughout the Ca²+ recording to ensure continuous CGRP receptor blocking. All results are presented as the mean ± standard error of the mean (SEM). *p < 0.05, ****p < 0.0001. Statistics performed by unpaired t-test.
CGRP binding and Piezo1 channel activation promote cellular remodelling via paxillin focal adhesion point reorganization
To assess and visualize cellular remodelling taking place upon exposure to either Yoda1 and/or CGRP, we stained the cells for FAs (paxillin) and actin microfilaments. Paxillin was frequently observed at the ends of actin filaments following stimulation with Yoda1 and/or CGRP; however, this pattern was not consistently present across all cells (Figure 5(A)). Quantitative analysis showed that paxillin FA points per cell were increased after treatment with Yoda1 and CGRP 1h_Yoda1 compared to the control condition (Figure 5(B)). The number of paxillin FA points was comparable between the immediate CGRP + Yoda1 exposure and the CGRP 1h_Yoda1.

Paxillin focal adhesion (FA) numbers and area size detection after Yoda1, CGRP, CGRP + Yoda1 and CGRP 1h_Yoda1 application in hiPSC-ECs. (A) Representative images of paxillin (red) immunocytochemical staining, Actin (green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue). Third row represents the images from the analysis after paxillin detection. Scale bar, 100 µm. (B) Average number of paxillin FA points per cell. (C) First row: zoom-in images of paxillin from (A). Second row: detected paxillin FA points in the range of 12–15 µm2 in the images from first row highlighted with yellow by ImageJ. Scale bars, 40 µm. (D) Average area of paxillin FA points per cell. Control: n = 6, DMSO: n = 6, Yoda1: n = 6, CGRP: n = 6, CGRP_Yoda1: n = 6, CGRP 1h_Yoda1: n = 6 (n = number of images analysed). All results are presented as the mean ± standard error of the mean (SEM). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistics performed by one-way analysis of variance with Tukey's multiple comparisons test.
To further evaluate the size of paxillin FAs, we analysed the area of paxillin-positive FA points (Figure 5(C)). As expected, in the lower area range (0–6 µm²), the number of paxillin FA points remained comparable across all treatment conditions. Within the range 6–12 µm2, neither Yoda1 nor CGRP alone influenced the FA area, whereas the combined treatment of CGRP and Yoda1 significantly increased the number of FA points compared to the control condition. Notably, CGRP 1h_Yoda1 treatment resulted in a greater number of FA points with larger areas (12–15 µm2) compared to all other treatments and the control condition (Figure 5(D)). Although the total number of FA points did not differ between immediate CGRP application and one hour CGRP exposure (Figure 5(B)), prolonged CGRP exposure led to a significant increase in FA area (Figure 5(D)). This finding underscores the impact of CGRP sensitization on Piezo1 activation.
Discussion
In the present study, hiPSC-ECs were exposed to CGRP in the presence or absence of Piezo1 channel activation (by applying agonist Yoda1) to investigate the crosstalk between these two migraine-related signalling pathways. The findings may offer preliminary insight into how mechanical and neuropeptide signalling in the vasculature could contribute to migraine pathophysiology. We show that, when Piezo1 channel agonist Yoda1 and CGRP were applied separately, both the intracellular Ca2+ level and the number of hiPSC-ECs responding increased compared to the respective control conditions. As can be expected, the combinations of CGRP + Yoda1 and CGRP 1h_Yoda1 lead to a significant increase in the number of cells responding compared to application of Yoda1 or CGRP alone. This can be attributed to the fact that two downstream pathways were targeted (Piezo1 and CGRP), and thus more cells respond to stimulation. The increased number of responding cells upon combined stimulation could potentially be explained by underlying heterogeneity in the expression or signalling efficiency of Piezo1 channels and CGRP receptors across the cell population. This is supported by our immunostaining results that showed heterogeneous expression of Piezo1 channels.
We report that Yoda1 and CGRP increase intracellular Ca2+ level in hiPSC-ECs. While Yoda1-induced Piezo1 channel activation leading to increased Ca2+ has been reported in human umbilical vein endothelial cells (HUVECs), this has not been studied in differentiated iPSCs. hiPSC-derived cells offer more personalized medicine applications compared to HUVECs because they can generate various cell types from a single individual (29–31). Previous research also showed that CGRP modulates endothelial function by enhancing Ca2+ levels, although mostly in animal models (32,33). Interestingly, combining CGRP + Yoda1 did not significantly change intracellular Ca2+ signals, suggesting that more cells respond with higher Ca2+ peaks, but the intensity threshold remains unaffected. We also examined the intracellular Ca2+ signal after one hour of CGRP sensitization followed by Piezo1 channel activation, aiming to model an aspect of migraine attacks. This aligns with reports that headache onset occurs 40–60 minutes after CGRP infusion (34). Indeed, CGRP 1h_Yoda1 significantly increased intracellular Ca2+ compared to other treatments. Testing CGRP or media after sensitization showed that only secondary Yoda1 application after CGRP sensitization led to a significant increase in Ca2+ level, supporting the hypothesis of crosstalk between CGRP and Yoda1. This can be further supported by the recently published paper from Zhang et al. (17), where they showed that Piezo1 channel activity is regulated by PKA phosphorylation at the S1612 site in response to stimuli like shear stress and GPCR agonists (e.g. CGRP), increasing Piezo1 sensitivity to mechanical changes. It could be that the one hour of CGRP exposure accumulates higher levels of PKA involved in the activation of Piezo1 channels. More specifically, it was demonstrated that PKA activation via 8-BrcAMP enhances Yoda1-induced Ca2+ influx in HUVECs, aligning with our findings (17). Additionally, PKC, which is part of the PLC pathway triggered by CGRP, can mediate Piezo1 channel activation, offering another route for CGRP-Piezo1 crosstalk (10,35).
After analysing Ca2+ intensity, we assessed the time to reach peak Ca2+ signal. CGRP leads to a faster Ca2+ peak than Yoda1, revealing two subgroups: early (<60 seconds) and late responders. This may reflect heterogeneous CGRP receptor expression or dual pathway activation. While CGRP primarily activates the cAMP/PKA pathway, it can also stimulate the PLC pathway via Gαq signalling, releasing Ca2+ from the ER. PLC activation, occurring alongside cAMP production, may cause delayed Ca2+ responses due to additional steps (e.g. IP3 binding) (10,36,37). Some cells may activate cAMP/PKA first, while others favor the PLC pathway, influenced by the spatial organization of CGRP receptors and signalling molecules (38). Additionally, CGRP 1h_Yoda1 induces a significantly faster Ca2+ peak than immediate CGRP + Yoda1, suggesting CGRP sensitization lowers Yoda1's mechanical activation threshold. Yoda1-induced Piezo1 channel opening may be potentiated by prior PKA/PKC accumulation, explaining the faster responses with CGRP 1h_Yoda1 compared to acute Yoda1 application. Notably, there was no significant difference in the time to peak Ca²+ signal between CGRP alone and CGRP 1h_Yoda1 groups. This can be explained by the lowered Yoda1 activation threshold after CGRP one-hour sensitization, resulting in fast and less variable Ca²+ responses that resemble those induced by CGRP alone, which are also driven by PKA/PKC accumulation. This suggests that CGRP 1h_Yoda1 effectively normalizes the slower Yoda1 responses via intracellular kinase-mediated sensitization, highlighting a key mechanistic interaction in calcium signalling.
To confirm that the response from hiPSC-ECs is due to CGRP, we measured intracellular Ca2+ level after applying BIBN4096, a CGRP receptor antagonist (39). BIBN4096 significantly reduces both the Ca2+ peak and the number of responding cells after CGRP application, confirming successful CGRP antagonism. This aligns with previous findings showing reduced Ca2+ level in nerve-mast cells after BIBN4096 treatment (40). Given its clinical relevance in migraine research, we also tested its effect on CGRP 1h_Yoda1 treatment, where it significantly decreases Ca2+ level, suggesting potential inhibition of vasodilation. Taken together, our findings lead us to hypothesize that the highest increase in Ca²+ signal following one hour of CGRP with subsequent Piezo1 channel activation may involve indirect crosstalk between CGRP and Piezo1 signalling pathways. However, further studies are needed to confirm this interaction and clarify the underlying mechanisms. The particularly high burst of Ca2+ can potentially be used by endothelial cells to produce higher levels of NO, which can be diffused and act on nearby smooth muscle cells, orchestrating downstream pathways that lead to vasodilation (41,42).
Piezo1 is known to influence FA dynamics (43–45), consistent with our finding that Yoda1 and CGRP 1h_Yoda1 increase paxillin FA points per cell compared to controls. In contrast, CGRP alone and CGRP + Yoda1 did not affect FA point numbers, indicating Piezo1's role in FA reorganization during cellular remodelling. Given that the intracellular Ca2+ level influences FAs, we also analysed FA size. Interestingly, CGRP 1h_Yoda1 increased paxillin FA size compared to other treatments, suggesting greater paxillin accumulation. It is known that the size of FAs reflects the strength and stability of cell adhesion (22). It has previously been shown that cellular force exertion on the ECM increases with FA size (46) and that increased cellular force can contribute to ECM stiffening (23). In light of those findings, we propose that the larger FA areas observed following CGRP 1h_Yoda1 treatment may be associated with enhanced force transmission to the ECM. While our data do not directly measure ECM stiffness, this potential increase in mechanical output could contribute to vascular stiffening. Such stiffening may reduce vessel wall compliance, thereby amplifying the activation of mechanosensitive nociceptors in response to pulsatile flow. This mechanism could underlie the localized pain sensation associated with CGRP release during migraine attacks. Further studies are required to directly test this hypothesis and to elucidate the causal relationship between FA remodelling and changes in vascular mechanical properties.
Conclusions
Here, we present an alternative 2D in vitro platform to investigate CGRP and Piezo1 mechanisms in hiPSC-ECs, serving as a first step towards more complex 3D neurovascular models of migraine. We demonstrated that one hour of sensitization of these cells with CGRP leads to increased Piezo1 activation, and together to a particularly high level of intracellular Ca2+ as well as elevated cellular remodelling, as evidenced by an increased number and thicker FA points in hiPSC-ECs. Our findings demonstrate that BIBN4096 effectively inhibits intracellular Ca²+ levels within this sensitization protocol, consistent with its known role as a potent CGRP pathway blocker. Importantly, this inhibition confirms that the effects observed after CGRP application are specific to CGRP receptor activation, supporting the specificity of our model system for studying CGRP signalling dynamics. We suggest that the observed cellular changes may be indicative of endothelial activation and cytoskeletal remodelling, which could be relevant to vascular tone regulation. The increased focal adhesion thickness might reflect localized changes in cell–matrix interactions that could influence local vessel mechanics. Such alterations in vascular behaviour may be relevant in the context of neurovascular contributions to migraine pathophysiology.
Article highlights
Establishment of an in vitro hiPSC-ECs cellular model to study migraine. CGRP one-hour sensitization enhances Piezo1 activation, causing increased intracellular Ca²+ levels and thicker FA points in hiPSC-ECs, which may reflect structural remodelling with potential implications for vascular mechanical properties. BIBN4096 inhibits CGRP-induced increase of intracellular Ca²+ in hiPSC-ECs. The findings suggest a link between mechanosensitive cellular changes and migraine-associated pain.
Supplemental Material
sj-png-1-cep-10.1177_03331024251404478 - Supplemental material for Indirect crosstalk between signalling pathways activated by CGRP and Piezo1 in human iPSC-derived endothelial cells relevant to migraine
Supplemental material, sj-png-1-cep-10.1177_03331024251404478 for Indirect crosstalk between signalling pathways activated by CGRP and Piezo1 in human iPSC-derived endothelial cells relevant to migraine by Vasiliki Gkouzioti, Ali Abdollahzadeh, Francijna van den Hil, Valeria Orlova, Rashid Giniatullin, Arn M. J. M. van den Maagdenberg, and Jean-Philippe Frimat in Cephalalgia
Supplemental Material
sj-png-2-cep-10.1177_03331024251404478 - Supplemental material for Indirect crosstalk between signalling pathways activated by CGRP and Piezo1 in human iPSC-derived endothelial cells relevant to migraine
Supplemental material, sj-png-2-cep-10.1177_03331024251404478 for Indirect crosstalk between signalling pathways activated by CGRP and Piezo1 in human iPSC-derived endothelial cells relevant to migraine by Vasiliki Gkouzioti, Ali Abdollahzadeh, Francijna van den Hil, Valeria Orlova, Rashid Giniatullin, Arn M. J. M. van den Maagdenberg, and Jean-Philippe Frimat in Cephalalgia
Supplemental Material
sj-png-3-cep-10.1177_03331024251404478 - Supplemental material for Indirect crosstalk between signalling pathways activated by CGRP and Piezo1 in human iPSC-derived endothelial cells relevant to migraine
Supplemental material, sj-png-3-cep-10.1177_03331024251404478 for Indirect crosstalk between signalling pathways activated by CGRP and Piezo1 in human iPSC-derived endothelial cells relevant to migraine by Vasiliki Gkouzioti, Ali Abdollahzadeh, Francijna van den Hil, Valeria Orlova, Rashid Giniatullin, Arn M. J. M. van den Maagdenberg, and Jean-Philippe Frimat in Cephalalgia
Footnotes
Acknowledgements
Author contributions
VG, JPF and RG designed the in vitro experiments. VG, LH and JPF performed and analysed the in vitro experiments. AA and VG performed the focal adhesion analysis. VG, RG, AMJMvdM and JPF interpreted the in vitro data. VG, JPF, RG, AA, LH, VO and AMJMvdM revised the manuscript.
Data availability
The data used for this article may be obtained from the authors upon reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: this work was supported by the Netherlands Organ-on-Chip Initiative, Gravitation project (024.003.001 to AMJMvdM) funded by the Ministry of Education, Culture and Science of the government of the Netherlands; the ZonMw funded JPND Grant “REBALANCE” (nr JPND2022–100; to AMJMvdM); and Research Council of Finland (grant #360360 to AA). ZonMw, Research Council of Finland, Ministerie van Onderwijs, Cultuur en Wetenschap, (grant number JPND2022-100, 360360, 024.003.001).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.