
Abstract
Background
Rimegepant is an orally administered small molecule calcitonin gene-related peptide receptor antagonist indicated for the acute and preventive treatment of migraine.
Methods
Two single-center, phase 1, open-label, randomized bioequivalence studies were conducted in healthy adult non-smokers, aged 18–55 years. One study compared the rate and extent of absorption of the marketed formulation of rimegepant 75 mg orally disintegrating tablet (ODT) administered sublingually with rimegepant 75 mg oral tablet, an earlier development formulation; the second compared the rate and extent of absorption of 75 mg rimegepant ODT administered supralingually with rimegepant oral tablet.
Results
The ln-transformed geometric mean ratios for the area under the curve (AUC) from time 0 to the last available concentration time point (time t) (AUC0-t), AUC from time 0 to infinity (AUC0-inf), and maximum observed concentration (Cmax) of sublingual rimegepant ODT vs. rimegepant tablet were 97, 97, and 105%, respectively, and the 90% confidence intervals (CIs) were all within the predefined range (80–125%) for bioequivalence. The ln-transformed geometric mean ratios for the AUC0-t and AUC0-inf of supralingual rimegepant ODT vs. rimegepant tablet were 98%, the 90% CIs were within the predefined range (80–125%), and the geometric mean ratio for Cmax was 103% with the 95% upper confidence bound for the scaled average bioequivalence criterion of –0.0575 (within-participant coefficient of variation for the reference for Cmax > 30%) for bioequivalence.
Conclusions
Rimegepant 75 mg ODT, administered sublingually or supralingually, and rimegepant 75 mg oral tablet were bioequivalent.
Introduction
When offered a choice between delivery systems for acute treatment, more than 90% of patients with headache conditions prefer oral formulations to parenteral and intranasal formulations, citing ease of use and fewer side effects as main rationale for their choice (1). At the same time, because of the added convenience and perception of a rapid onset of action with orally disintegrating tablet (ODT) formulations, patients with migraine have been shown to prefer them to traditional oral tablets (2–4). Following oral administration, ODTs are rapidly disintegrated and absorbed without the need for additional fluid, which facilitates easy administration of acute treatment to patients with migraine. In the treatment of migraine, this benefit may be especially important to the many patients who experience nausea before and during attacks who wish to avoid drinking liquids.
Rimegepant is an orally administered small molecule calcitonin gene-related peptide (CGRP) receptor antagonist that has demonstrated efficacy in the acute and preventive treatment of migraine (5–9). A previous dose-ranging trial with rimegepant determined that the optimal dose in migraine, 75 mg, provided relief that was comparable to sumatriptan 100 mg (10). Two oral formulations of rimegepant have been evaluated in phase 3 trials: an ODT (6,9) that is the marketed formulation and an earlier development formulation of a conventional oral tablet (5,7,8). The pharmacokinetic (PK) profiles of rimegepant ODT and oral tablet have not previously been directly compared, nor has their bioequivalence been ascertained. It is also unknown whether the mode of oral administration of rimegepant ODT (i.e. sublingual vs. supralingual) affects PK parameters relative to conventional oral administration. Accordingly, these studies were conducted to compare the rate and extent of absorption of rimegepant ODT administered sublingually and supralingually with rimegepant oral tablet, as well as to assess the safety, tolerability, and PK of rimegepant ODT and oral tablet, in healthy, fasted volunteers.
Methods
Ethics
These studies adhered to the ethical principles of the Declaration of Helsinki and were conducted in accordance with Good Clinical Practice, as defined by the International Council on Harmonisation Harmonized Tripartite Guideline, and applicable regulatory requirements. The protocols were reviewed and approved by the Institutional Review Board for the study center. Before any study-related procedures were undertaken, participants were clearly and fully informed about the purpose, potential risks, and other critical issues regarding the studies, and participants provided their informed consent.
Design
Both of these methodologically identical studies were single-center, phase 1, open-label, randomized four-period, two-sequence, fully-replicated crossover bioequivalence studies in healthy, fasted adults. In one, study treatments were sublingual rimegepant 75 mg ODT and rimegepant 75 mg oral tablet, each administered twice; in the second, study treatments were supralingual rimegepant 75 mg ODT and rimegepant 75 mg oral tablet, each administered twice. In both studies, participants were confined to the research facility from day −1, at least 10 hours before the first rimegepant dose, until the morning of day 22, the duration of study participation was approximately 3 weeks, and the interval between administrations of study medication was at least 5 days.
Participants
Eligible participants included males and females who had not used tobacco products within 3 months prior to screening and who were aged at least 18 years and not more than 55 years, with a body mass index greater than 18.5 kg/m2 and less than 30.0 kg/m2, and a body weight of at least 50.0 kg for males and 45.0 kg for females. Participants also had to have no clinically significant illness or surgery within 4 weeks prior to dosing; no history of neurological, endocrinal, cardiovascular, pulmonary, hematological (e.g. neutropenia), immunologic, psychiatric, gastrointestinal, renal, hepatic, or metabolic disease; and a score of 0 at screening on the Sheehan Suicidality Tracking Scale (11) (S-STS), a 16-question, patient self-reported or clinician-administered rating scale that can be used to track treatment-emergent suicidal ideation and behaviors. Participants also had to be able to understand the nature of the study, agree to comply with the prescribed dosage regimens, and communicate to study personnel about adverse events (AEs) and concomitant medication use, as applicable. Female participants were required to have a negative serum pregnancy test prior to dosing with study medication. Sexually active males and females of childbearing potential were required to use effective birth control methods for at least 60 days after receiving the last dose, and males were not allowed to donate sperm throughout the study and for at least 90 days after receiving the last dose of investigational product.
Participants were excluded if they had a current diagnosis of viral hepatitis, a history of liver disease or a positive test for hepatitis B antigen, hepatitis C virus, or human immunodeficiency virus during medical screening; significant history of seizure disorder other than a single childhood febrile seizure (e.g. epilepsy); or a current or recent (within 3 months of study drug administration) gastrointestinal disease, including gastrointestinal surgery that might interfere with physiological absorption and motility. They could not participate if they had used medication (other than topical products without significant systemic absorption) or any drugs known to induce or inhibit hepatic drug metabolism within 30 days prior to the first study drug administration. Participants with any clinically significant deviation from normal in physical examination, vital signs, electrocardiogram (ECG), or clinical laboratory determinations beyond what was consistent with the target population were also ineligible.
Treatments
In both studies, fasting participants (no food from ≥10 hours predose until ≥4 hours postdose and no fluids, except for water given with standard oral tablet, from 1 hour predose until 1 hour postdose) were administered each of two treatments twice. In one study, the test treatment was rimegepant 75 mg given as a single ODT to be held under the tongue (sublingually) until fully disintegrated then swallowed without water; the reference treatment was rimegepant 75 mg given as a single standard oral tablet swallowed with water. In the second study, the test treatment was rimegepant 75 mg given as a single ODT to be held on the top of the tongue (supralingually) until fully disintegrated then swallowed without water; the reference treatment was rimegepant 75 mg given as a single standard oral tablet swallowed with water. In both studies, treatments were administered on day 1 during each of the four periods, with a washout of at least 5 days between doses.
Assessments
Pharmacokinetics
Primary PK endpoints in both studies included area under the curve from time 0 to the last available concentration time point (time t) (AUC0-t), area under the curve from time 0 to infinity (AUC0-inf), and maximum observed concentration (Cmax) calculated using rimegepant plasma data. Secondary PK endpoints were time of Cmax (Tmax), elimination rate constant (Kel), elimination half-life (T1/2el), and residual area.
In both studies, 68 blood samples (17 for each of the four treatments) were drawn from each participant for PK analyses. They were collected prior to drug administration and 5, 10, 20, 30, 40, and 50 minutes and 1, 1.5, 2, 2.5, 5, 8, 12, 24, 48, and 72 hours postdose (3 ml for each sampling time; time tolerance window ±29 seconds). Sample collections done outside the predefined time windows were not considered protocol deviations because actual postdose sampling times were used for PK and statistical analyses. Unless otherwise specified or for participant safety, when blood draws and other procedures coincided, blood draws had precedence. A dead-volume intravenous catheter was used for blood collection to avoid multiple skin punctures, when appropriate. Otherwise, blood samples were collected by direct venipuncture.
Safety
Both studies evaluated the safety and tolerability of rimegepant through the assessment of AEs, S-STS determinations, local tolerability assessment of the ODT, clinical laboratory parameters, 12-lead ECGs, vital signs, and physical examinations.
Laboratory assessments included biochemistry (albumin, alkaline phosphatase, aspartate aminotransferase, alanine aminotransferase, gamma glutamyl-transpeptidase, urea, calcium, chloride, glucose, phosphorus, potassium, creatinine, sodium, creatine phosphokinase, direct bilirubin, indirect bilirubin, total bilirubin and total protein); serology (hepatitis B surface antigen, hepatitis C virus antibody, and human immunodeficiency virus antigen and antibody); and hematology (complete blood count with differential, hemoglobin and hematocrit). Other safety assessments included coagulation (only after abnormal — i.e. >3× the upper limit of normal (ULN) liver function tests) and urinalysis (macroscopic examination, pH, specific gravity, protein, glucose, ketones, bilirubin, occult blood, nitrite, urobilinogen and leukocytes).
Physical examinations, vital signs, and physical measurements (blood pressure, respiratory rate and sitting heart rate) were evaluated at screening and at study exit. Oral temperature was measured at screening and at study exit. Blood pressure, heart rate, and oral temperature were measured before dosing on day 1 and 24, 48, and 72 hours after each treatment. When vital signs measurements coincided with a blood draw, they were performed before the blood collection whenever possible. Vital signs performed at study exit were used, as the vital signs required 72 hours postdose after the last treatment. Body measurements were performed at screening and included body weight, height measurement, and body mass index.
A supine 12-lead ECG was performed at screening, before dosing on day 1 in each period, and at study exit. When an ECG coincided with a blood draw, it was performed before the blood collection whenever possible. A urine pregnancy test was performed at screening and at study exit. A serum pregnancy test was performed at check-in.
In both studies, participants receiving rimegepant ODT had a local tolerability assessment with oral cavity inspection at screening, on day 1 before dosing, approximately 15 minutes after dosing in each period, and at study exit. Alterations of the tongue, palate, or buccal mucosa were recorded as AEs.
Other
The S-STS was administered by study personnel and completed on site at screening and at study exit. If investigators determined that a participant was at risk of suicide or self-harm, appropriate measures to ensure the participant’s safety and obtain mental health evaluation were taken, and the participant was discontinued from the study. The event was recorded as either an AE or serious AE as determined by the investigator and reported to the sponsor within 24 hours.
Randomization
Participants were treated according to a four-period, two-sequence block randomization scheme in both studies. The computer-generated randomization codes were not available to study staff until the clinical and analytical phases of the study were complete. Because of the objective nature of the data, these studies were open-label; treatment assignments were randomized, but there was no blinding.
Sample size
Based on the results of a preliminary study (unpublished data), the intraparticipant coefficient of variation in both studies was expected to be approximately 30% for AUC and Cmax. With this expected coefficient of variation and an expected ratio of AUC and Cmax within 0.91 and 1.10, the study had a power of at least 80% to show bioequivalence with 30 participants in a four-period fully replicated design. To maintain analytical power while accounting for dropouts, at least 36 participants were included.
Statistical analysis
Both studies defined the safety population as all participants who received at least one dose of the study medication. They defined the PK populations as all participants completing at least two periods without major protocol violation and for whom the PK profile could be adequately characterized.
Pharmacokinetics
In both studies, the PK parameters of rimegepant were derived by non-compartmental analysis from individual plasma vs. time profiles using Phoenix WinNonlin, version 6.4 (Certara, Princeton, NJ, USA). Inferential statistical analyses of log-transformed AUC0-t, AUC0-inf, and Cmax were performed using the MIXED Procedure in SAS, version 9.4 (SAS Institute, Cary, NC, USA). According to Food and Drug Administration (FDA) requirements (12,13), if the within-participant coefficient of variation for the reference (CVWR) of rimegepant tablet was less than 30% for a primary parameter (AUC0-t, AUC0-inf or Cmax), then the 90% confidence interval (CI) for the ratio of geometric means (ODT/tablet), based on least squares means from the analysis of variance of the log-transformed data, had to be within 80–125% to conclude bioequivalence for that parameter. For any primary parameter with CVWR greater than or equal to 30%, the scaled average bioequivalence approach was applied as follows: the point estimate of the test/reference ratio had to be within 80–125%, and the 95% upper confidence bound for the scaled average bioequivalence criterion had to be ≤0 to conclude in favor of bioequivalence for that parameter. Untransformed Tmax, Kel, and T1/2el were analyzed using PROC MIXED Procedure at an alpha level of 0.05. The bioequivalence analyses were also performed according to European Medicines Agency (EMA) requirements (14) and also included the Wilcoxon signed-rank test on the median Tmax for both treatments.
Safety
Safety data were not subjected to inferential analysis.
Results
Participants
The duration of the sublingual study was 90 days, with the first participant enrolling on 18 November 2017 and the last participant completing on 16 February 2018. The supralingual study lasted 117 days; the first participant enrolled on 26 August 2018 and the last participant completed on 20 December 2018.
In the sublingual study, 84 participants were screened, 48 (57%) were enrolled, and 35 (73%) received at least one dose of rimegepant; in the supralingual study, 121 were screened, 65 (54%) were enrolled, and 36 (55%) received at least one dose of rimegepant (Table 1). All treated participants in both studies (sublingual 35/35, 100%; supralingual 36/36, 100%) received the first administration of rimegepant ODT and tablet. In the sublingual study, 34 (97%) participants received the second administration of rimegepant ODT and tablet; in the supralingual study, 32 (89%) received the second administration of study treatments. The safety population in the sublingual study included 35 participants and the PK population included 34 participants. The safety and PK populations in the supralingual study included 36 participants.
Disposition of participants in the studies comparing rimegepant 75 mg ODT administered sublingually or supralingually with rimegepant 75 mg oral tablet.
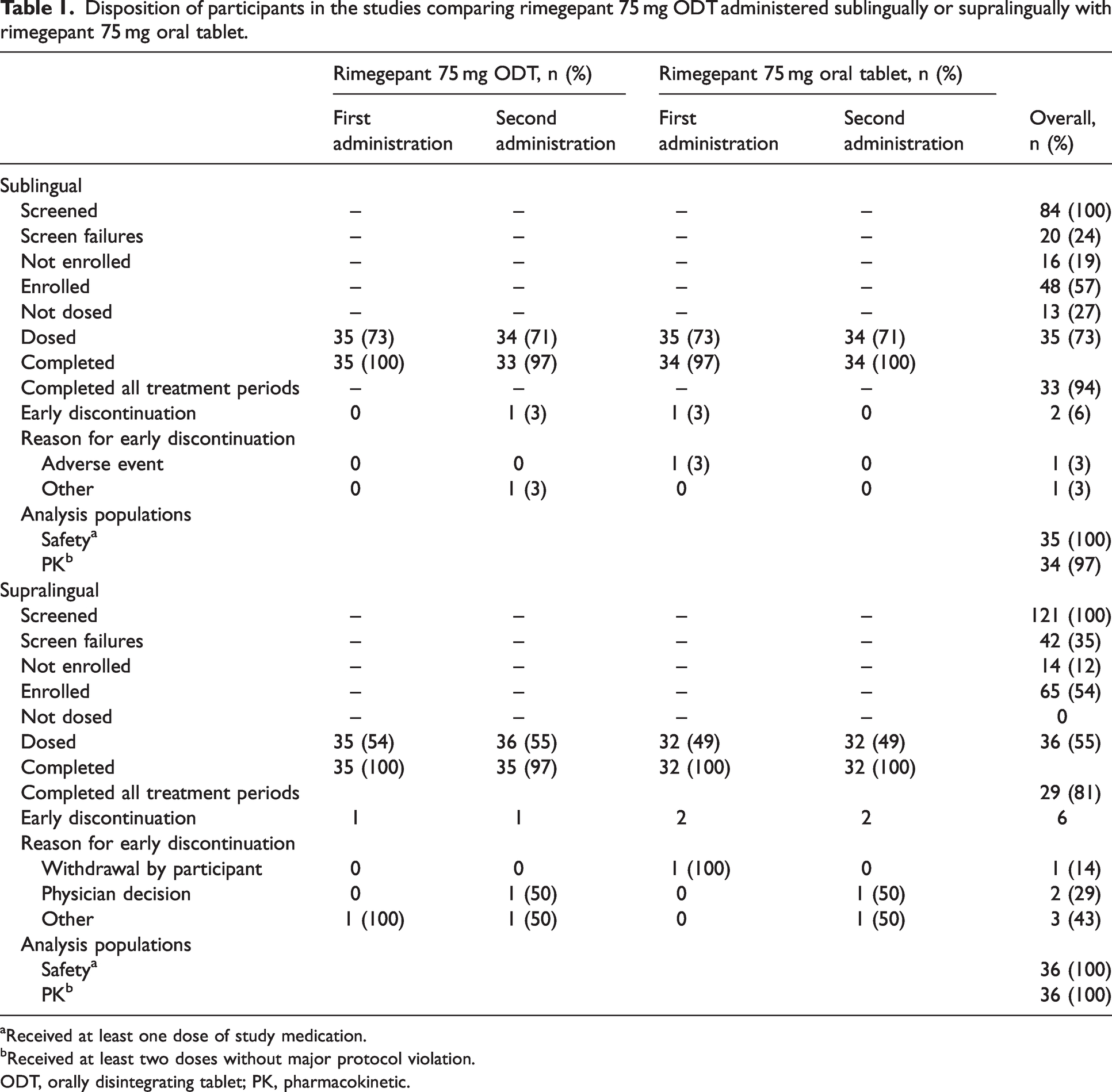
Received at least one dose of study medication.
Received at least two doses without major protocol violation.
ODT, orally disintegrating tablet; PK, pharmacokinetic.
Demographics and other baseline characteristics of the study populations are presented in Table 2. In the sublingual study, participants (N = 35) had mean (SD) age of 37.7 (9.5) years; 80% (28/35) were male, and 80% (28/35) were white. Mean (SD) body mass index was 25.85 (2.4) kg/m2. In the supralingual study, participants (N = 36) had mean (SD) age of 39.5 (9.4) years; 53% (19/36) were male and 94% (34/36) were white. Mean (SD) body mass index was 25.25 (2.5) kg/m2.
Demographics and other baseline characteristics in the studies comparing rimegepant 75 mg ODT administered sublingually or supralingually with rimegepant 75 mg oral tablet.

Pharmacokinetics
The rimegepant plasma concentration vs. time curves from the sublingual study are presented in Figure 1. The PK parameters are summarized in Table 3. In the sublingual study, the CVWR values for AUC0-t, AUC0-inf, and Cmax were all less than 30%. The geometric mean ratios for the AUC0-t, AUC0-inf, and Cmax of sublingual rimegepant 75 mg ODT vs. rimegepant 75 mg tablet point estimates were 97, 97, and 105%, respectively, with 90% CI within the predefined range (80–125%) for bioequivalence according to FDA requirements (Table 4). Bioequivalence for rimegepant sublingual ODT and oral tablet were also met according to EMA requirements. The Tmax for sublingual rimegepant ODT was 29 minutes earlier than for the oral tablet, with median (range) untransformed rimegepant Tmax values of 1.50 (0.66–2.50) hours for the ODT and 1.99 (0.66–5.03) hours for the tablet (p = 0.0028 based on the Wilcoxon signed-rank test).

Median rimegepant plasma concentrations in participants treated with sublingual rimegepant 75 mg ODT and rimegepant 75 mg oral tablet on (a) linear and (b) semi-log scales.
Summary of the pharmacokinetics of rimegepant 75 mg ODT and rimegepant 75 mg oral tablet administered sublingually or supralingually.

ODT vs. oral tablet p=0.0028 (Wilcoxon signed-rank test).
ODT vs. oral tablet p=0.2408 (Wilcoxon signed-rank test).
CV, coefficient of variation; Max, maximum; Min, minimum; ODT, orally disintegrating tablet.
Bioequivalence of rimegepant 75 mg ODT administered sublingually or supralingually and rimegepant 75 mg oral tablet per FDA requirements.

Calculated using least squares means according to: e(DIFFERENCE) × 100.
Geometric confidence interval unless specified otherwise.
Reference-scaled average bioequivalence approach.
AUC, area under the curve; CI, confidence interval; Cmax, maximum observed concentration; CVWR, within-participant coefficient of variation for the reference; ODT, orally disintegrating tablet.
The rimegepant plasma concentration vs. time curves from the supralingual study are presented in Figure 2 and the PK parameters are shown in Table 3. In the supralingual study, the CVWR values for AUC0-t and AUC0-inf were less than 30%, but the CVWR for Cmax was greater than 30%. The geometric mean ratios for the AUC0-t and AUC0-inf of supralingual rimegepant 75 mg ODT vs. rimegepant 75 mg tablet point estimates were both 98%, with 90% CI within the predefined range (80–125%) for bioequivalence. The point estimate of the geometric mean ratio for Cmax was 103%, within the range 80–125%, and the 95% upper confidence bound for the scaled average bioequivalence criterion was −0.0575 (≤0), in favor of bioequivalence according to FDA requirements (Table 4). Bioequivalence for supralingual rimegepant ODT and oral tablet was also met according to EMA requirements. There was no statistical difference between the Tmax values for supralingual ODT and the oral tablet, and the median (range) untransformed rimegepant Tmax values were 2.00 (0.66–5.02) hours for the ODT and 2.05 (0.66–5.01) hours for the tablet (p = 0.2408 based on the Wilcoxon signed-rank test).

Median rimegepant plasma concentrations in participants treated with supralingual rimegepant 75 mg ODT and rimegepant 75 mg oral tablet on (a) linear and (b) semi-log scales.
Safety
In the sublingual study, 35 participants were analyzed for safety, and 17 (49%) experienced treatment-emergent AEs (TEAEs), as presented in Table 5. The only AEs reported by more than one participant were constipation (n = 6; 17%); increased level of alanine transaminase (n = 3; 9%; none >2.1× ULN); and heart rate increased, headache, nasopharyngitis and back pain (each n = 2; 6%). No severe or serious AEs were reported. There were four TEAEs of moderate intensity: nausea, headache, hypotension, and otitis externia.
Treatment-emergent adverse events occurring in more than one participant following rimegepant 75 mg ODT administered sublingually or supralingually and rimegepant 75 mg oral tablet.

ALT, alanine aminotransferase; ODT, orally disintegrating tablet; TEAE, treatment-emergent adverse event.
In the supralingual study, of the 36 participants analyzed for safety, 13 (36%) participants experienced TEAEs (Table 5). The only AEs reported by more than one participant were headache (n = 6; 17%), back pain (n = 3; 8%) and nasopharyngitis (n = 2; 6%). No severe or serious AEs were reported. There was one TEAE of moderate intensity: headache.
No clinically meaningful changes from baseline in laboratory values, vital signs, ECGs, or the S-STS were observed in either study.
Discussion
These randomized, four-period, two-sequence crossover bioequivalence studies in healthy, fasted participants were conducted to evaluate the bioequivalence of rimegepant 75 mg ODT and oral tablet, as well as to assess the safety, tolerability, and PK of both rimegepant forms. The bioequivalence of sublingual and supralingual rimegepant ODT with rimegepant oral tablet was demonstrated by finding, in both studies, that the 90% CIs for the ratio of geometric means for log-transformed AUC0-t, AUC0-inf, and Cmax were between 80 and 125%. The Tmax of the sublingual ODT occurred 29 minutes earlier than the oral tablet; there was no significant difference in Tmax in the supralingual study. In both studies, single doses of rimegepant 75 mg ODT or tablet were well tolerated, and the safety profiles of the both forms were comparable, with no clinically significant findings on the local tolerability or oral safety assessments, no deaths or serious AEs reported, and no clinically meaningful changes from baseline in laboratory values, vital signs, ECGs, or the S-STS.
The favorable safety results observed in these studies are consistent with findings from previous separate assessments of the ODT and oral tablet forms of rimegepant, which identified no safety concerns and found that the tolerability of rimegepant was comparable to placebo (5,7,9,10).
With nausea a feature of every migraine attack in 32% of patients and most migraine attacks in 56% of patients, as well as the given reason for delaying or avoiding treatment by 31% of patients (15), a treatment option that does not involve swallowing a tablet or water, such as the ODT, may encourage some patients to treat earlier in the attack, improving compliance and thereby outcomes in clinical practice.
Some acute treatments for migraine have been developed in ODT form and demonstrated acceptable efficacy, although their PK profiles are inferior to the oral tablet forms. For example, rizatriptan ODT and zolmitriptan ODT have demonstrated efficacy in clinical trials (16–19) and have published preference studies showing that patients with migraine prefer the ODT forms to oral tablets because they perceive them to be easier to use, better tolerated in the presence of nausea, faster, and more effective (3,4,20). Yet rizatriptan ODT administered without water has a lower AUC0-2h than the oral tablet (18.83 vs. 30.03 hours·ng/ml), and both rizatriptan ODT and zolmitriptan ODT have substantially later median Tmax values than the oral tablets: 1.33 hours vs. 0.67 hours for rizatriptan (21) and 3 hours vs. 1.5 hours for zolmitriptan (22). Other ODT forms of migraine drugs have been developed, although the efficacy of these ODT forms has not yet been reported, such as sumatriptan (23), frovatriptan (24), a fixed combination of naproxen and naratriptan (25), and ondansetron (26).
Rimegepant 75 mg ODT, sublingually or supralingually, demonstrated bioequivalence with rimegepant 75 mg oral tablet. The results of these bioequivalence studies led to two phase 3 randomized controlled trials in adults with migraine (6,9), which showed that rimegepant 75 mg ODT was more effective than placebo. Importantly, the demonstrated bioequivalence between the marketed rimegepant ODT and the development formulation oral tablet bridges the results of clinical studies using the oral tablet (5,7,8) with those using the ODT (6,9) and supports the approval of rimegepant 75 mg ODT for both acute and preventive treatment of migraine.
These bioequivalence studies have some limitations. They were conducted in healthy volunteers and outside the context of a migraine attack and therefore did not address the intraictal PK profile of rimegepant. In addition, when the ODT was administered sublingually in one of the studies, the relative amounts of rimegepant that were absorbed sublingually, orally, and swallowed could not be determined.
Conclusions
In healthy, fasted adults, rimegepant 75 mg ODT administered sublingually or supralingually and rimegepant 75 mg oral tablet were bioequivalent. The ODT and oral tablet forms of 75 mg rimegepant were similarly well tolerated.
Clinical implications
Two single-center, phase 1, open-label, randomized, four-period bioequivalence studies were conducted to compare the rate and extent of absorption of rimegepant 75 mg ODT administered sublingually or supralingually with rimegepant 75 mg oral tablet and to assess the safety, tolerability and PK of both formulations in healthy, fasted adults. In both studies, rimegepant 75 mg ODT, administered sublingually or supralingually, and rimegepant 75 mg oral tablet were bioequivalent. In both studies, rimegepant 75 mg ODT and rimegepant 75 mg oral tablet were well tolerated, with no severe or serious AEs reported.
Footnotes
Acknowledgments
Medical writing services were provided by Christopher Caiazza at Polymedia Corporation and funded by Pfizer.
Data availabilty
Declaration of conflicting interests
Robert Croop was an employee of Biohaven Pharmaceuticals, owns stock in Biohaven Ltd, was an employee of Pfizer, has received research payments from Pfizer and provides services to Collima LLC, which has had consulting agreements with Pfizer, Aptose Biosciences Inc., Manistee Therapeutics and Vida Ventures Management Co., L.L.C. Jennifer Hould and Richard Bertz are employed by and own stock/stock options in Biohaven Pharmaceuticals. Jing Liu and Kyle T. Matschke are employed by and own stock/stock options in Pfizer Inc. Rajinder Bhardwaj, PhD, and Matt S. Anderson, PhD, are employed by Certara USA, which was a paid consultant of Biohaven Pharmaceuticals. Richard B. Lipton serves on the editorial board of Neurology and as senior advisor to Headache but is not paid for his roles on these journals. He has received research support from the NIH. He also receives support from the National Headache Foundation. He receives research grants from Allergan/Abbvie, Amgen, Dr Reddy’s Laboratories and Novartis. He has reviewed for the NIA and NINDS and serves as consultant, advisory board member, or has received honoraria from Allergan/Abbvie, Amgen, Biohaven, Dr Reddy’s Laboratories, electroCore, Eli Lilly, GlaxoSmithKline, Merck, Novartis, Teva and Vedanta. He receives royalties from Wolff’s Headache (8th edition, Oxford University Press, 2009) and Informa. He holds stock options in Biohaven Pharmaceuticals and Manistee.
Ethical statement
This study adhered to the ethical principles of the Declaration of Helsinki and was conducted in accordance with Good Clinical Practice, as defined by the International Council on Harmonisation Harmonized Tripartite Guideline, and applicable regulatory requirements. The protocol was reviewed and approved by the Institutional Review Board at the study center. Before any study-related procedures were undertaken, participants were clearly and fully informed about the purpose, potential risks and other critical issues regarding the study, and participants provided their informed consent.
Funding
This study was funded by Biohaven Pharmaceuticals, which was acquired by Pfizer in October 2022.