
Abstract
Background
This narrative review aims to discuss several common neurological and psychiatric disorders that show comorbidity with migraine. Not only can we gain pathophysiological insights by studying these disorders, comorbidities also have important implications for treating migraine patients in clinical practice.
Methods
A literature search on PubMed and Embase was conducted with the keywords “comorbidity”, “migraine disorders”, “migraine with aura”, “migraine without aura”, “depression”, “depressive disorders”, “epilepsy”, “stroke”, “patent foramen ovale”, “sleep wake disorders”, “restless legs syndrome”, “genetics”, “therapeutics”.
Results
Several common neurological and psychiatric disorders show comorbidity with migraine. Major depression and migraine show bidirectional causality and have shared genetic factors. Dysregulation of both hypothalamic and thalamic pathways have been implicated as a possibly cause. The increased risk of ischaemic stroke in migraine likely involves spreading depolarizations. Epilepsy is not only bidirectionally related to migraine, but is also co-occurring in monogenic migraine syndromes. Neuronal hyperexcitability is an important overlapping mechanism between these conditions. Hypothalamic dysfunction is suggested as the underlying mechanism for comorbidity between sleep disorders and migraine and might explain altered circadian timing in migraine.
Conclusion
These comorbid conditions in migraine with distinct pathophysiological mechanisms have important implications for best treatment choices and may provide clues for future approaches.
Introduction
There are prevailing theories about migraine pathophysiology, foremost that cortical spreading depolarization (CSD) is the mechanism underlying the migraine aura and that activation of the trigeminovascular system causes the headache (1). The exact mechanisms for attack initiation are still unknown, which complicates the identification of novel treatment targets. Several neurological and psychiatric disorders are comorbid with migraine, and some increase the chance of migraine chronification. These comorbidities may provide clues for shared pathophysiological mechanisms, either through direct causal relationships or through shared underlying genetic and environmental factors. For this review we selected highly prevalent neurological and psychiatric comorbidities that have important clinical impact, are well-researched, and have the potential to increase our understanding of migraine. As such, this review explores the most recent advances in understanding the comorbidity of migraine with mood disorders, ischemic stroke, epilepsy and sleep disorders. We highlight clues about shared underlying mechanisms and brain regions (Figure 1), with an emphasis on genetic risk factors. While clinical trials on treating migraine with comorbidities are scarcely available, we focus on possible treatment consequences and practical advice that health-care providers taking care of migraine patients should be aware of.

Brain regions most involved in the comorbidity of migraine with depression, stroke, epilepsy and sleep disorders.
Migraine and mood disorders
Epidemiology
Depression and anxiety are both associated with an increased risk of migraine, which is even higher when both co-occur in a patient (2). Around 45–65% of patients with a depressive disorder meet the criteria for an anxiety disorder, vice versa 30–65% of patients with an anxiety disorder also have depression (3). Comorbidity of migraine and depression is bidirectional. Lifetime prevalence of depression is three times higher in migraine patients and having migraine constitutes higher risk for onset of depression (relative risk [RR] ∼1·5) (4). Migraine prevalence in patients with bipolar disorder is also increased, with estimates ranging from ∼30–50% (5).
A direct causal effect of pain on depressive symptoms seems plausible. However, major depression signals higher risk for first occurrence of migraine (odds ratio [OR] ∼3·4), but not for other headaches (6). Yearly, 3% of patients convert from episodic to chronic migraine (≥15 headache days/month, of which ≥8 migraine days) (7). Depressive symptoms and allodynia during attacks and acute medication overuse are interlinked and lead to chronification (8).
Pathophysiological mechanisms
Thalamic pathways may be implicated in migraine attack initiation foremost in central sensitization during attacks, which is experienced as allodynia (9). Decreased thalamic activity has been reported in migraine with comorbid depression, compared to migraine and (major) depression separately, implicating a habituation deficit (10). Diverse neurochemical pathways converge on thalamic neurons, including noradrenergic, dopaminergic and serotonergic pathways implicated in depression, depicting an intersection of pathways for migraine. A magnetic resonance spectroscopy (MRS) study supported this suggested thalamocortical dysfunction in migraine chronification, as evidenced by bilateral thalamic N-acetyl-aspartate reductions (11). Although migraine and depression pathophysiology involves monoamine neurotransmitters, how these neurotransmitters contribute towards disease needs further elucidation (12). In the past most pathophysiological explanations concerning migraine and depression comorbidity focused on serotonin (5-HT). For instance, 5HT1a receptor density was increased in migraine on positron emission tomography (13), but decreased in depression (14). Regardless, it is now widely accepted that additional neurotransmitter mechanisms are involved.
Premonitory and ictal symptoms in migraine such as nausea and yawning supposedly involve the hypothalamus. The observation that both migraine and bipolar disorder are associated with an evening chronotype, suggests involvement of the circadian system pointing towards hypothalamic involvement (5), and might suggest a role for dopamine. While dopamine is a major neurotransmitter involved in depression, its importance in migraine is still disputed (12).
Alternatively, depression is hypothesized to involve dysregulation of the adaptive stress response, an innate mechanism aimed at restoring homeostatic level altered by chronic stress. Eventually, this might progress to multiple systemic pathologies such as inflammation and a prothrombotic state potentially resulting in development of co-morbid conditions such as migraine (15).
Shared genetic susceptibility
Migraine and mood disorders show familial occurrence, with complex inheritance and a combination of genetic and environmental factors together conferring disease susceptibility. In genome-wide association studies (GWAS) many susceptibility loci have been identified for migraine and depression (16,17). Combining large GWAS provided evidence for genetic overlap for these disorders (cross-disorder genetic correlation (rG = 0·25)) (18), while pointing at neural-related pathways of signaling and ion channel (dys)regulation. A small study found that a family history of bipolar disorder is associated with an increased risk of migraine (OR ∼4·4), suggesting a shared genetic basis, but could not establish an increased risk of migraine in those with a family history of major depressive disorder (19). Several mood disorders, including major depressive disorder and bipolar disorder, show shared genetic etiology (20). In the Brainstorm Consortium collaboration, the genetic sharing of 25 brain disorders (>250,000 patients, >750,000 controls) were quantified based on GWAS (21). While neuropsychiatric disorders showed considerable sharing of common variants, neurological disorders appeared more distinct from one another and from the psychiatric disorders, except for migraine that revealed a shared genetic basis with depression (rG = 0·32) (21). A possible explanation for the former is that clinical neuropsychiatric disease features show considerable overlap, more than for neurological disorders.
Epidemiological and genetic studies suggest that migraine with aura patients have a higher risk of developing depression compared to patients suffering from migraine without aura (OR ∼1·7) (22). Increased risk was found for hemiplegic migraine (OR ∼3·7), a subtype with motor auras (23). Hemiplegic migraine can be caused by mutations in genes encoding ion transporters subunits, CACNA1A, ATP1A2 and SCN1A and functional characterization of these mutations point to neuronal and glial dysfunction, resulting in enhanced glutamatergic neurotransmission (24). Of note, studies using in vivo proton MRS reported decreased levels of glutamate metabolites in regions of the medial frontal cortex in depression (25), while an increase was demonstrated in the visual cortex in migraine (26).
Shared environmental factors
Exposure to stress, primarily regulated by corticotropin-releasing factor (CRF)-containing neural circuits and the hypothalamic–pituitary–adrenal (HPA) axis, can precipitate depression. Stress is a frequently self-reported migraine trigger (27), but there is little proof for abnormal stress response in migraine patients (12). Perceived acute stress might instead be part of the premonitory phase, which is characterized by other symptoms such as tiredness and food cravings (28). As such, premonitory symptoms can be misidentified as triggers. Depression is a major determinant of perceived stress in migraine patients (29). Migraine, depression and medication overuse form a complex triad. Depression is a risk factor for developing medication overuse headache (8). Withdrawal from medication is an efficacious treatment for patients with chronic migraine and medication overuse (30). However, worldwide there is debate on whether to withdraw first or add preventive treatment. Patients with comorbid depression are often excluded from such studies as they are more treatment resistant.
Consequences for clinical practice
Medication overuse is often implicated in patients with comorbid depression and chronic migraine. Withdrawal of all acute medications should be considered first choice of treatment, ideally supported by a headache nurse (31,32). When initializing prophylactic treatment, screening for comorbid depression is recommended. This is because prophylactic drugs such as topiramate, flunarizine and beta-blockers may provoke depressive symptoms (33), whereas valproate may have mood-stabilizing effects. Anti-depressant drugs may be effective against both depressive symptoms and migraine (Table 1). Amitriptyline might be particularly effective when patients also have tension-type headache; nortriptyline has not been studied systematically. Selective serotonin reuptake inhibitors (SSRIs) or selective norepinephrine reuptake inhibitors (SNRIs) for migraine prophylaxis seem not sufficient (34). Risk of serotonin syndrome in patients taking SSRI or SNRI and using triptans, serotonin agonists for acute treatment in migraine, is low (35). Lithium is often the first treatment for preventing mood instability in bipolar disorder. Interestingly, a history of migraine was associated with failure to respond to lithium (36). Finally it is important to realize that the presence of depression might influence a patient’s response to treatment. One such example is Onabotulinumtoxin A, in which a poorer response was associated with depressive symptoms (37). Recently, calcitonin gene-related peptide (CGRP) monoclonal antibodies together with small-molecule antagonists have become available as anti-migraine drugs (38). An increasing number of studies have suggested that the presence of depression or depressive symptoms was associated with a poorer response to anti-CGRP monoclonal antibodies (39,40). However, not all studies found this association (41). Moreover, in a post-hoc analysis anti-CGRP preventive treatment appeared effective in preventive treatment of chronic migraine in patients with comorbid depression compared with placebo (42).
Recommendation regarding anti-migraine treatment and comorbidities.
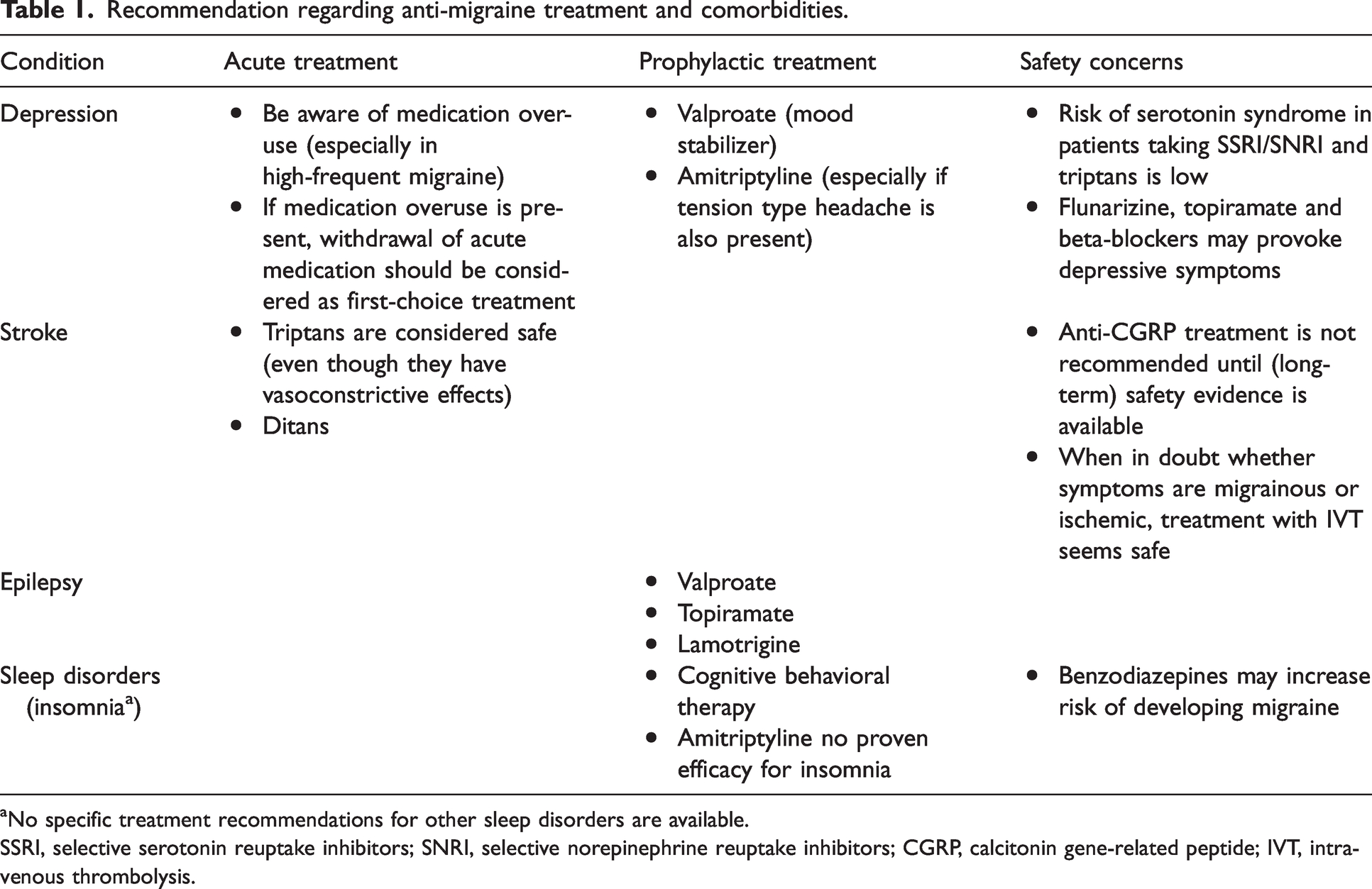
No specific treatment recommendations for other sleep disorders are available.
SSRI, selective serotonin reuptake inhibitors; SNRI, selective norepinephrine reuptake inhibitors; CGRP, calcitonin gene-related peptide; IVT, intravenous thrombolysis.
Migraine and stroke
Epidemiology
Migraine doubles the risk for ischemic stroke, most evidently for migraine with aura (43). Migraine may increase individual vulnerability to ischemic stroke during acute brain ischemia (44), for hemorrhagic stroke this is controversial (45). Migraine can be a stroke risk factor or might be an (early) symptom of underlying structural abnormalities (46). Population-based studies showed increased white matter hyperintensities and posterior lesions in migraine (47). Although progressive over time lesion development occurs independently of migraine attack frequency (47), and total lesion load remains limited (48).
Pathophysiological mechanisms
The mechanism underlying migraine aura is cortical spreading depolarization (CSD), a wave of neuronal and glial cell depolarization that propagates over the cortex, followed by a prolonged phase of nerve cell depression (1), associated with hypoperfusion (49). This neurovascular mechanism may be the link between migraine and stroke. Although CT-perfusion scans are mostly normal in migraine with aura patients that present to the emergency room as a stroke mimic, the aura symptomatology in these patients has often already resolved (50). Importantly, in patients with prolonged (motor) aura symptoms perfusion defects have indeed been described (51).
Animal studies have shown that micro-emboli can trigger SDs, even without causing micro-infarctions (52). This may explain why right-to-left cardiac shunting, often due to a patent foramen ovale (PFO), is a risk factor for stroke, but also associated with migraine with aura (53). SD has also been shown to affect ischemic stroke development, with peri-infarct depolarizations (the functional equivalent of SD in a migraine aura) circling around and causing expansion of the infarct core (54). Transgenic hemiplegic migraine mouse models with experimentally induced stroke had larger infarcts due to a higher propensity and more profound impact of peri-infarct depolarizations (55). This implies an increased tissue sensitivity to a supply-demand mismatch. Interestingly, migraine prophylactics were able to protect mice from ischemic injury (54). Clinical studies investigating this hypothesis, however, have not shown consistent results (44,56,57).
Another possibly shared mechanism for migraine and stroke is endothelial dysfunction, which is associated with an increased rate of cerebro- and cardiovascular events (58). The endothelium is a single layer of endothelial cells that line the interior surface of blood vessels. It acts as a barrier between the blood and extravascular space, but is also a major player in many processes, among which are regulating immunological processes, inflammation and angiogenesis, controlling blood fluidity, aggregation of platelets and vascular tone, and finally it acts by functioning as a metabolizing and endocrine organ. Endothelial dysfunction has been implicated in migraine, with reduced cerebrovascular reactivity occurring in migraine patients (59). Markers for endothelium activation, hypercoagulability, and inflammation are also associated with migraine (58). Systemic inflammation with endothelial involvement may be associated with migraine and stroke, as is seen in cases with systemic lupus erythematosus or other auto-immune disorders (60). Enhanced platelet aggregation has also been demonstrated in patients with migraine (61,62), and while interesting, additional well-designed studies are needed to determine if this mechanism, possibly controlled by the endothelium, provides the link between migraine and stroke on the cellular level. Reduced vascular reactivity itself might also (partly) explain the link between migraine and stroke. A recent meta-analysis demonstrated more arterial stiffness and impaired cerebral vasodilator function, demonstrating altered cerebrovascular function (63).
Shared genetic susceptibility
Several cerebral hereditary angiopathies are associated with migraine and cerebrovascular disease, such as Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic manifestations (RVCL-S), COL4A1- and COL4A2-associated diseases, Dutch-type Cerebral Amyloid Angiopathy (D-CAA), and Mitochondrial Encephalopathy, Lactic acidosis, And Stroke-like episodes (MELAS) (Figure 2) (64). Less convincing evidence is available for common genetic variants, like the variants described in the MTHFR and ACE D/I genes (64). Most clearly a shared genetic susceptibility of migraine and stroke is suggested by GWAS findings (65), including the identification of a shared locus for migraine and large artery strokes that includes the LMOD2-WASL gene region (65). Of note, WASL is implicated in stabilizing endothelial adherens junctions (66), and fits the hypothesis that endothelial dysfunction seems involved.
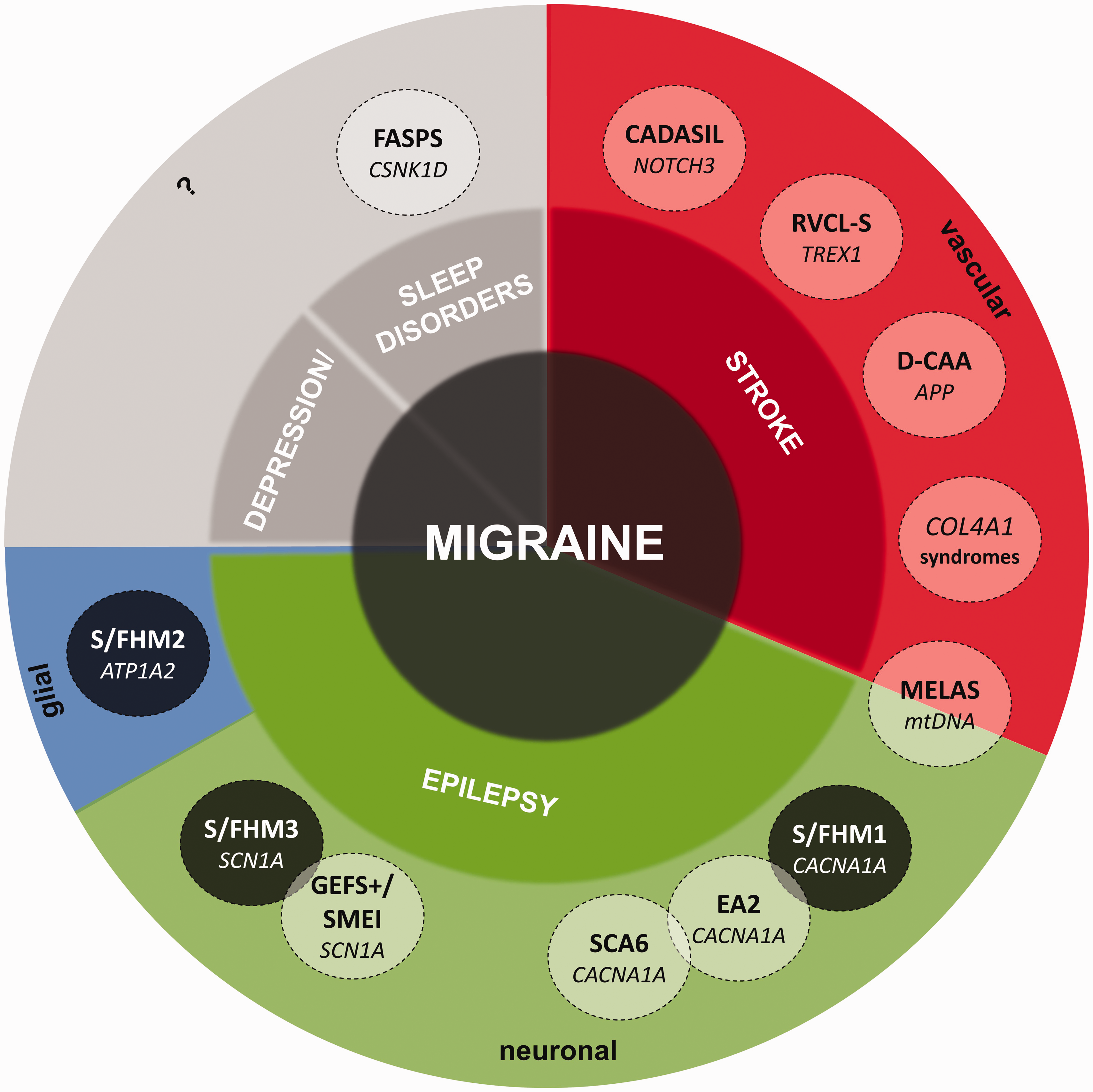
Monogenic syndromes associated with migraine and comorbidity with stroke, epilepsy, depression and sleep disorders. Depicted are relations of these monogenic syndromes with migraine and each other, causative genes (in italics) and the main associated pathophysiological pathways: Sporadic and Familial Hemiplegic Migraine type 1, 2 and 3 (
Regardless, no significance at the individual patient level can be attributed to these genetic findings, nor is screening for polymorphisms useful in clinical practice. Still, loci at LRP1, a member of the LDL receptor family, were implicated in migraine, stroke and cervical artery dissection, suggesting a shared biology that deserves further study (67). Moreover, through a GWAS approach single nucleotide polymorphism (SNP) rs9349379 in the PHACTR1 locus (6p24) was implicated as risk factor for five vascular diseases including migraine (68). Sophisticated (epi)genetic analyses that also involved induced pluripotent stem cells, indicated a regulatory function of this SNP on the expression of the distantly (600 Kb) located endothelin 1 (EDN1) gene. Although it is puzzling that this DNA variant through endothelin increases the risk of cardiovascular disease whereas it decreases migraine risk, this type of analysis illustrates nicely that integration of phenotypic and (epi)genetic data can be a promising avenue to identify potential disease mechanisms.
Mendelian randomization (MR) is a research method that may provide insight of causal relations between risk factors and disease. Recent MR studies did not show direct causal relationships between cardiovascular risk factors and migraine but instead evidence of horizontal pleiotropy, meaning that stroke, migraine, and cardiovascular risk factors share SNPs as genetic factors (69–71).
Shared environmental factors
There is an increased stroke risk in (older) smokers with migraine (hazard ratio [HR] ∼3·2), compared with non-smokers (HR ∼0·8) (72). Oral contraceptive use in female migraine patients results in a ∼8-fold increased risk of ischemic stroke, which increases to an up to >30-fold increased risk with concurrent smoking in female migraine with aura patients (73). In many guidelines, migraine with aura is consequently considered a contra-indication for oral contraceptives, although this may not be true for new oral contraceptives with lower estrogen doses. While absolute risks of stroke are still low, smoking and combined use of oral contraceptives should be advised against in women with migraine with aura. Migraine was shown not to be associated with atherosclerosis in large vessels in patients with acute ischemic stroke (74). In contrast, 1H-NMR metabolite profiling of 225 lipoproteins in plasma samples from large cohorts showed decreased levels of HDL-associated lipoproteins (apoA1, S-HDL-FC ratio, omega-3 fatty acids) to be associated with migraine (75). Finally, it is suggested that migraine frequency may decrease after a stroke, presumably due to vascular or cortical brain changes, change in medication or lifestyle, or PFO closure (76).
Consequences for clinical practice
Because of vasoconstrictive effects, triptans have been contraindicated in migraine patients with cardio- or cerebrovascular disease. Notably, in observational studies no increased stroke risk was found, although those prescribed a triptan might have had more favorable vascular risk profiles (77). Nonetheless, triptan users with cardiovascular medication (as proxy for higher vascular risk) did not show an increased stroke risk (78). Based on expert opinion, triptan use nowadays is considered safe in stroke patients in many countries, and might even be used in migraine-stroke disorders such as CADASIL. The recently developed ditans (selective 5-HT1F receptor agonists) do not cause vasoconstriction, and may have preference over triptans (79).
CGRP monoclonal antibodies and small-molecule antagonists are also important to consider (38). CGRP is released from neurons in the trigeminal ganglion and binds to receptors on meningeal vessels, causing vasodilation and neurogenic inflammation (38). Blocking vasodilation caused by CGRP as escape response to an ischemic event may potentially lead to a larger area of infarction (80). So far, subjects with higher vascular risk were excluded from clinical trials (38), and long-term adverse events will have to be awaited. In vitro studies demonstrated that erenumab significantly inhibited CGRP-mediated vasodilation (while not interacting with other vasoactive compounds) (81) and that fremanezumab also had direct functional effects on CGRP-mediated vasodilation (82). Furthermore, work in mice demonstrated that treatment with small molecule CGRP antagonists led to worsening of cerebral ischemic outcomes (83). A handful of studies have evaluated the effect of erenumab in vivo and while one demonstrated that erenumab did not alter vasomotor reactivity or flow-mediated dilation in migraine patients without aura, another demonstrated that it did affect trigeminovascular reactivity (84,85). Further large in vivo studies are needed to determine in more detail how the vasculature of patients with migraine responds to these drugs. As hypertension is a known risk factor for ischemic stroke, it is important to note that real world data already indicated that treatment with anti-CGRP (receptor) antibodies led to an increase in mean systolic and diastolic blood pressure, and that some of the treated patients required antihypertensive treatment for the increase in blood pressure (86,87). Blood pressure is a major modifiable risk factor for stroke over a broad range of blood pressure (88). As such, physicians should monitor the blood pressure of patients when they are treated with anti-CGRP (receptor) antibodies. Furthermore, a report of an ischemic stroke in a 41-year-old woman, using rizatriptan and oral contraceptives, without pre-existing cardiovascular comorbidities following administration of a CGRP receptor blocker, (89) may raise concerns and it seems warranted to exclude patients with significant vascular risk factors from use of anti-CGRP treatment until evidence of long-term safety is available (Table 1). As such, stroke should be considered a contraindication until the long-term use of monoclonal antibodies targeting CGRP or its receptor are further investigated.
Acute stroke treatment increasingly consists of intravenous thrombolysis (IVT). In 10% of patients treated with IVT clinical follow-up reveals stroke-mimics, among them migraine (with aura), in whom IVT treatment fortunately seems safe (90). PFO closure in stroke patients is debated, although in stroke patients with a migraine history, PFO prevalence may be up to 80% (91). It is highly debatable whether migraine frequency improves after PFO closure, with studies reporting conflicting results (92). New onset of migraine has been reported within days after PFO or atrial septal defect closure, with improvement after antiplatelet treatment (93). Other studies suggested that treatment with acetylsalicylic acid might be effective as migraine prophylaxis for migraine with aura (94). Most studies were of limited methodological quality and the possibly effective dose is unclear. Therefore, acetylsalicylic acid is not recommended as migraine prophylaxis.
Migraine and epilepsy
Epidemiology
Lifetime prevalence of migraine is increased in epilepsy patients (OR ∼1·5) and, vice versa, lifetime prevalence of epilepsy is increased in migraine patients (OR ∼1·8) (95). The comorbidity is particularly frequent in hemiplegic migraine, with epilepsy occurring in ∼40% of hemiplegic migraine patients (96). Migraine aura-triggered seizures may also develop during or directly following (<60 minutes) a migraine with aura attack, which has not been described in migraine without aura (7). Ictal epileptic headache is a rare phenomenon during focal seizures that is not necessarily migrainous but may also fit characteristics of tension-type headache (7). Both entities have been referred to as ‘migralepsy’, a term advised to avoid because of its unclear meaning. Post-epileptic headache, which may have migrainous features, is more common, most frequently after bilateral tonic clonic seizures (7). Epilepsy patients also suffering from migraine have a worse prognosis, with lower probability of becoming seizure-free (OR ∼0·3) and increased risk of treatment-resistance (OR ∼6·2) (97).
Pathophysiological mechanisms
Neuronal hyperexcitability is considered a shared causal mechanism for migraine and epilepsy (98). This mechanism may cause CSD initiation in migraine, while in epilepsy this may lead to simultaneous firing of populations of cortical neurons. CSD events may even show epileptic-like electrophysiological characteristics, with recordings of CSD evolving into epileptic convulsions (99). Possibly, CSD causes synchronization in the affected neurons, which may lower the threshold for epileptic seizures (100). (Hyper)synchronization during epileptic seizures may also prime the brain for CSD. On the other hand, the repolarization phase or depression following epileptic seizures or CSD may also raise the threshold for one another, indicating that co-occurrence of attacks is dependent on timing of pathophysiological processes.
Shared genetic susceptibility
A genetic link between migraine and epilepsy lies in the overlapping involvement of genes encoding ion transporters in hemiplegic migraine and epilepsy, i.e. CACNA1A, ATP1A2, SCN1A, and PRRT2 (64). Hemiplegic migraine patients with the p.Ser218Leu CACNA1A mutation, for instance, suffer from a particularly severe phenotype, that includes seizures, cerebral edema and coma that can be fatal (101). Transgenic Cacna1aS218L mice exhibit increased susceptibility to induced CSD (102). Homozygous Cacna1aS218L mice display severe attacks, spontaneous (fatal) seizures, and serve as a promising animal model for sudden unexpected death in epilepsy (SUDEP) (103). During seizures, spreading depolarizations in the mutant brainstem are found, which cause respiratory and subsequent cardiac arrest and death. The beneficial effects of NMDA receptor blockers hold the promise for targets to prevent this dramatic course (104).
One of the ATP1A2-associated familial hemiplegic migraine type 2 (FHM2) mouse models shows increased susceptibility to CSD and epileptiform activity (105), similar to patients with the same p.Gly301Arg ATP1A2 mutation (106). A higher susceptibility to CSD correlates with severe epileptiform activity possibly related to increasing extracellular potassium (105). Apart from the ATP1A2-encoded Na+K+-ATPase pump α2 subunit, the excitatory amino acid transporters (EAAT1 and EAAT2) are implicated in this clearance, and constitute therapeutic targets for migraine and epilepsy.
The SCN1A gene is implicated in FHM3 and epilepsy syndromes, including Dravet syndrome/severe myoclonic epilepsy of infancy (SMEI) and generalized epilepsy with febrile seizures (GEFS+) (107). Spontaneous CSD events were shown in transgenic FHM3 mice expressing the p.L263V SCN1A mutation (108). Gain-of-function of NaV1.1 channels seems to result in hemiplegic migraine whereas loss-of-function leads to epilepsy (107). Possibly, FHM3 SCN1A mutations lead to accumulation of potassium, while epileptogenic SCN1A mutations facilitate depolarization block of GABAergic neurons, thereby both increasing neuronal hyperexcitability, but through different pathophysiological mechanisms (109).
Mutations in the PRRT2 gene are mainly associated with paroxysmal kinesigenic dyskinesia, benign familial infantile seizures and infantile convulsion choreoathetosis syndrome, but have also been found in a few patients with (also) hemiplegic migraine, in which PRRT2 mutations may act as a genetic cofactor (110). PRRT2 encodes a proline-rich transmembrane protein. Mutations likely cause neuronal hyperexcitability by affecting neurotransmitter release (111).
For common migraine, an increased prevalence of migraine with aura was found in families containing two or more individuals with epilepsy of unknown cause, suggesting shared genetic susceptibility (112). A GWAS in epilepsy revealed 16 loci, the majority of which are associated with generalized epilepsy (113), suggesting a role for ion channel genes (SCN1A, SCN2A and SCN3A), and genetic correlations of epilepsy with migraine with aura specifically.
Shared environmental factors
Brain damage due to traumatic head injury or stroke may cause post-traumatic headache resembling migraine and increases the risk of epilepsy (7). Besides such direct causal factors, there is a large overlap in suggested triggers, such as stress, sleep deprivation, fatigue, alcoholic beverages, and hormonal changes (27,114). In rodents, sleep deprivation enhances CSD susceptibility (115). The influence of stress on CSD appears more complex. In FHM1 mice, corticosterone injection, but not restraint stress, led to enhanced CSD susceptibility (116), illustrating that corticosterone can enhance CSD susceptibility whereas the rising of corticosterone levels during acute (restraint) stress are not sufficient. Likely, stress gives rise to a more complex biological response with multiple positive and negative modulators. Interestingly, relief after chronic stress did reduce CSD threshold in FHM1 mice (117).
Consequences for clinical practice
It may be difficult to distinguish migraine aura from epileptic seizures, particularly when diagnosis is based on patient reports, symptoms are focal, or aura is not followed by headache. Positive visual symptoms can occur in both disorders, Todd’s palsies may be confused with hemiplegic migraine, and post-ictal headache may have migrainous characteristics. Time interval may be helpful, as aura symptomatology develops more gradually (minutes instead of seconds) in migraine. (Inter)ictal EEGs can be helpful. Routine EEGs cannot detect CSD but direct-current EEG might have a role in the future (118). Interestingly, patients with migraine and those with epilepsy have different antiepileptic drug adverse events profiles (119). A retrospective study in women with epilepsy demonstrated that valproate and topiramate are prescribed most frequently in those with comorbid migraine (120). When in doubt about whether a patient suffers from migraine, epilepsy, or both, prophylactic treatment can be directed at both disorders, with valproic acid, topiramate or lamotrigine (Table 1). Epilepsy patients with a confirmed SCN1A mutation should not be treated with sodium channel blockers, as this may worsen their clinical symptoms. Given the postulated gain-of-function of hemiplegic migraine-related SCN1A mutations, it is unclear whether effects of sodium channel blockers may in fact be beneficial in FHM3 patients.
Migraine and sleep disorders
Epidemiology
A variety of sleep disorders are suggested to be comorbid with migraine, including insomnia, restless leg syndrome (RLS), and obstructive sleep apnea. Odds ratios for insomnia in migraine patients vary between 1·5–2·5 (121). Insomnia shows a bidirectional relation with migraine, with insomnia preceding worsening of migraines and migraine being a precursor to insomnia (RR ∼ 1·7) (121). RLS prevalence ranges from 9–39% (due to heterogeneity of study designs) with ORs of 1·2–4 (122). Vice versa, migraine prevalence in RLS ranges between 15–63%, independent of sex or migraine type (122). Comorbidity of sleep apnea syndrome and migraine has been suggested, but evidence was lacking in a large population-based study (123). Snoring was two-fold more common among individuals with chronic daily headache than episodic headache, independent of traditional sleep apnea risk factors, suggesting an association with snoring rather than sleep apnea (124). In addition, as obesity is a risk factor for sleep apnea and likely also for migraine, this may also be the cause for the observed comorbidity (125).
Pathophysiological mechanisms
Migraine and RLS appear to have a temporal association, suggesting that one may trigger the other, with RLS triggering migraine explained by poorer sleep quality (122). Other mechanisms may involve dysfunction of the hypothalamic dopaminergic system. Dopamine-involvement in migraine is related to symptoms of yawning, food craving, drowsiness, mood changes, nausea and vomiting during either the prodromal, headache or postdromal phase of attacks (126). Interestingly, prodromal symptoms are more common in patients with comorbid RLS (126).
Chronobiological mechanisms play a role in migraine pathophysiology as attacks often start in early morning, and patients report being early chronotypes and less flexible in circadian rhythm changes (127). Serotonin and orexin/hypocretin systems are involved in the pathophysiology of migraine and sleep disorders, and these systems regulate pain perception and sleep–wake rhythm (128). The locus coeruleus, regulating the sleep-wake-cycle, is also involved in migraine pathophysiology. Dysfunctional locus coeruleus signaling has shown to alter attack susceptibility and to reduce CSD susceptibility threshold (129).
Sleep disorders can create an imbalance causing accumulation of glycogen during sleep (130). Glycogen is a vital energy reservoir required to match synaptic demand particularly for re-uptake of potassium and glutamate during glutamatergic transmission. Synaptic metabolic stress caused by insufficient glycogen-derived energy substrate can lower the CSD threshold (131). Sleep deprivation-induced changes may trigger migraine by reducing glycogen availability. Interestingly, chronic stress is suggested to increase glycogen turnover and decreased glycogen has been implicated in depression-like behavior in rats (132). Therefore, dysregulation of the glycogen system might link migraine to sleep disorders as well as depression.
Shared genetic susceptibility
Cannabinoid-type-1 receptors (CNR1 gene) are involved in sleep regulation, and Cnr1 knock-out mice show sleep alterations (133). Heterozygous rare coding variants in CNR1 were associated with headache, including migraine (134). Endocannabinoids may be a common underlying pathway for migraine and sleep disorders. MEIS1 variants were associated with increased risk of RLS in migraine, implicating an imbalance in iron homeostasis and the dopaminergic system (135). Finally, mutations in CSNK1D were reported in familial migraine and familial advance sleep phase syndrome (FASPS) (136).
Shared environmental factors
Migraine and sleep disorders may have overlapping trigger factors, such as stressful life events and working night shifts. Sleep deprivation is often reported as a migraine trigger and is associated with more frequent and severe migraine, a risk for migraine chronification (27,137). An experimental setting in mice showed that insufficient glycogen supply due to an administered glycogen phosphorylase inhibitor and sleep deprivation lowered the threshold for induced CSD (131).
Consequences for treatment in clinical practice
Use of antidepressants for insomnia is common. While it may seem an attractive ‘triple-hit’ option for patients with migraine, depression and insomnia, there is no evidence for amitriptyline use for insomnia (138). A small improvement in sleep quality was found for (short-term) use of doxepin and trazodone, but these drugs are not beneficial for migraine. With the adverse effects of antidepressants, such as daytime sleepiness, these drugs are not recommended for migraine patients suffering from insomnia. Short-acting benzodiazepines for insomnia may increase the risk of developing migraine (Table 1) (139). Cognitive-behavioral insomnia therapy does seem of benefit for (chronic) migraine patients with insomnia (140). Whether treatment of RLS also relieves migraine is speculative. Sleep apnea headache (7), usually recurs daily upon awakening and may be confused with chronic migraine. Screening patients with daily morning headache for sleep apnea is thus warranted, especially when other risk factors such as obesity are present.
Conclusion
Migraine shows comorbidity with several neurological and psychiatric disorders, with strongest and bidirectional relations with mood disorders and epilepsy. Migraine patients also have a higher risk for ischemic stroke. Several sleep disorders, most strikingly insomnia and RLS, also show bidirectional comorbidity with migraine.
Overlapping pathophysiological mechanisms are spreading depolarizations for migraine and stroke and hyperexcitability for migraine and epilepsy. For mood disorders and sleep disorders shared pathophysiological pathways are less clear, although (hypo)thalamic involvement and locus coeruleus involvement are suggested.
Studies on polygenic risk factors for migraine and its comorbidities have yielded limited results so far. It is not straightforward to link an associated SNP to the actual gene of effect. For GWAS to have true meaning in elucidating pathophysiological mechanisms, efforts should not only aim towards performing larger GWAS but also to designing ways to identify implicated pathways with functional studies. Monogenic syndromes show strong links between migraine and stroke as well as migraine and epilepsy and migraine and sleep disorders. Studying these disorders as models will provide important mechanistic insights. Epigenetic mechanisms, such as DNA methylation and histone modification, likely are also involved in migraine pathophysiology. Only small steps have been taken to unravel their contribution towards migraine (141), let alone towards understanding its comorbidities. Moreover, gene-environment interaction should also be evaluated as genetic factors modulating sensitivity to environmental exposures, or vice versa, might be equally important to understand the relationship between migraine and its comorbidities.
In most studies life-time diagnoses are used, regarding migraine and its comorbidities as traits. However, individuals with depression after the age of 50, are more likely to suffer from headache than younger people with the same risk factors (142). How differences in migraine subtype and frequency over lifespan may relate to its comorbidities is largely unknown. One can postulate that evolving environmental factors across lifespan as well as biological age, e.g., with changed sex hormone levels, may be important. The elderly also have age‐related renal and hepatic function changes that may alter drug metabolism and elimination, and the chances of multiple medication use and interactions are greater, which future studies should take into account.
Lastly, it is important to consider that migraine comorbidities might also be comorbid with one another, which might influence pathophysiological mechanisms and treatment options (143,144). In this light, it is interesting to realize that many psychiatric disorders display genetic overlap, while this is much less the case for neurological disorders. Here migraine was an exception, which showed significant genetic correlation with several psychiatric disorders (21).
Simultaneous treatment of comorbid disorders can be beneficial, regarding both drug treatment and tackling shared environmental risk factors, such as acute medication overuse, smoking, and sleep deprivation. Unfortunately, most clinical trials exclude participants with comorbid disorders, limiting evidence for optimal treatment for these patients. Nonetheless, assessing the presence of comorbid disorders may direct physicians towards other choices regarding treatment, not only to prevent harm but aim for positive effects on comorbidities. Altogether, neurological and psychiatric comorbidities do not only provide pathophysiological insight, they also have important implications for clinical practice.
Article highlights
Overlapping pathophysiological mechanisms between migraine and stroke are spreading depolarizations, while hyperexcitability is a key feature of both migraine and epilepsy. For mood disorders and sleep disorders shared pathophysiological pathways are less clear, although (hypo)thalamic involvement and locus coeruleus involvement are suggested. Simultaneous treatment of migraine and comorbid disorders can be beneficial, however, dedicated trials are needed.
Footnotes
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: N. Pelzer declares no conflicts of interest. I. de Boer reports research support by the Dutch Heart Foundation, Stichting Dioraphte and the International Retinal Research Foundation. A.M.J.M. van den Maagdenberg declares no conflicts of interest. G.M. Terwindt reports independent support from the Dutch Research Council, and the Dutch Hearth and Brain Foundations, Dioraphte and IRRF, she reports consultancy or industry support from Novartis, Lilly and Teva, Allergan (Abbvie), Lundbeck.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.