
Abstract
Background
Calcitonin gene-related peptide monoclonal antibodies (CGRP mAb) are an effective treatment of migraine however may have possible off-target effects. Pre-clinical studies implicate CGRP in several aspects of bone turnover and homeostasis. The clinical effect of CGRP mAb on bone turnover is not known, however.
Methods
Between June 2021 and July 2022, a multi-centre prospective cohort study was undertaken with eligible patients undergoing paired testing of the validated bone turnover markers procollagen type I N-terminal propeptide (P1NP) and serum C-terminal telopeptide of type I collagen (CTX) prior to and at least three months following administration of a CGRP mAb.
Results
A total of 45 patients with a mean age of 41.8 (SD 11.9) were included in the final analysis, all of whom received a ligand-targeting CGRP mAb. Administration of a CGRP mAb was associated with a statistically significant increase in P1NP from 44.5 microg/L to 51.5 microg/L (p = 0.004), but no significant change in CTX.
Conclusion
In otherwise homeostatic conditions, short-term administration of a CGRP mAb is associated with increased P1NP, a bone formation marker but not with increased CTX, a bone resorption marker. Further study is required to validate these findings over longer time periods, in a larger cohort, and in pre-existing states of increased calcium stress and bone-turnover.
Introduction
Calcitonin gene-related peptide (CGRP) is a 37 amino-acid peptide of the calcitonin family of peptides, with two isoforms (α-CGRP and β-CGRP) (1). Transcription of the CALCA gene exons 1–4 produces calcitonin mRNA; inclusion of exons 5–6 in place of exon 4 creates α-CGRP mRNA and β-CGRP is produced by a separate gene (2). Another peptide in the calcitonin family is amylin, which shares 40% amino acids with α-CGRP and structural similarities with both α-CGRP and calcitonin (3). Receptors for the calcitonin-family are comprised of varying components including calcitonin receptor (CTR), calcitonin receptor-like receptor (CLR) and receptor activity modifying proteins (RAMP) 1–3 (3). CLR and RAMP1 form the canonical CGRP receptor, CTR and RAMP1–3 form the amylin receptors and CTR forms the calcitonin receptor (3).
CGRP has emerged as an integral step in migraine genesis, and by inhibition of the CGRP ligand or receptor, an effective therapeutic target in both the prevention and acute treatment of migraine (1,4). CGRP receptors are expressed in multiple organ systems however, and have a wide range of described biological effects, leading to the postulation of off-target effects of CGRP inhibition (1). To date, there have been limited clinical reports of potential off-target effects including possible inflammatory complications and migraine-related stroke (5,6). A potential effect of CGRP inhibition on bone turnover has been postulated, but has not been studied clinically previously (1,7).
The homeostasis of bone turnover is an intricate system with multiple counter-balancing regulatory processes (8–10). Increasingly over the past two decades, it has been recognised that the central nervous system has a key role in the regulation of bone turnover (9). The emerging literature on this regulation includes the role of neural pathways such as the sympathetic nervous system, as well as neuropeptides such as the calcitonin family (9). The interaction between CGRP and bone turnover is summarised in Figure 1 and in detail below.
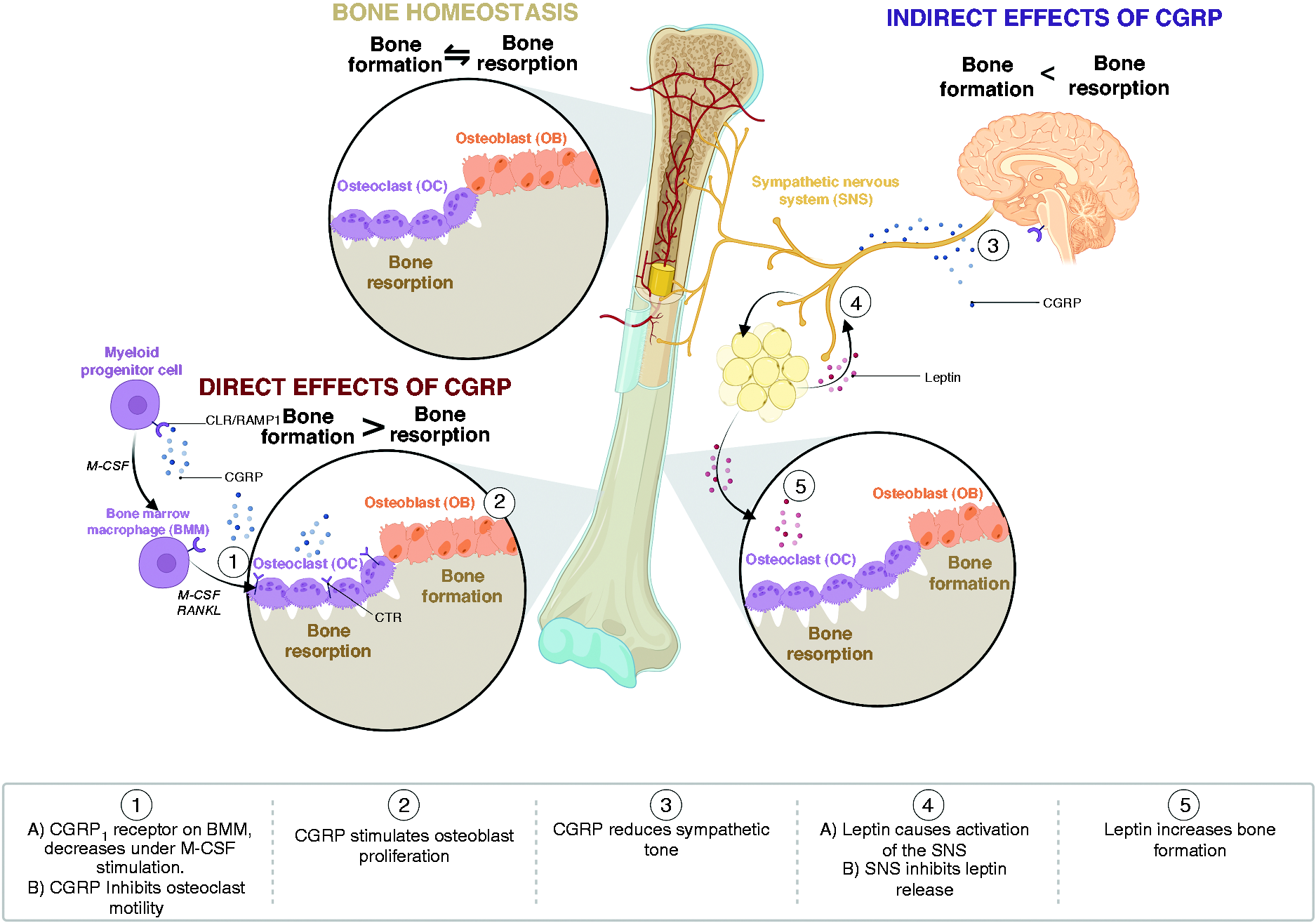
Summary of the pre-clinical evidence of Interaction between calcitonin gene-related peptide (CGRP) and bone turnover. Inhibition of CGRP would be presumed to have an inverse effect. BMM; bone marrow macrophage, CGRP; calcitonin gene-related peptide, CLR; calcitonin receptor-like receptor, CTR; calcitonin receptor, M-CSF; macrophage colony stimulating factor, RAMP1; receptor activity modifying protein 1, RANKL; receptor activator of nuclear factor κ-B ligand, SNS; sympathetic nervous system.
Direct interaction: Osteoclasts
Osteoclast differentiation, proliferation and activity are key in bone resorption. Myeloid progenitor cells differentiate into bone-marrow macrophages (BMM) under the stimulation of macrophage colony stimulating factor (M-CSF) before ultimately differentiating into osteoclasts under dual stimulation of M-CSF and receptor activator of nuclear factor-κB ligand (RANKL) (11).
Significantly, BMM in mice have been shown to express CLR and RAMP1 mRNA (the CGRP1 receptor), however under the stimulation of M-CSF, decreases of RAMP1 and increases of CTR mRNA (calcitonin receptor) were observed (12). In a pure population of osteoclast-like cells from co-cultures of bone marrow and spleen, RAMP1 and RAMP3 mRNA was undetectable (13). Taken together, this suggests that a direct effect of CGRP on osteoclast activity may only occur at the level of BMM.
Pre-clinically, CGRP, calcitonin and amylin all inhibit osteoclast motility by a receptor-based mechanism, and injection of the CGRP peptide has been shown in animals to lower plasma calcium levels, albeit in orders of magnitude less than calcitonin (14,15). Whether CGRP would have the same effect on osteoclast differentiation in vivo under the differential effect of M-CSF stimulation and resultant down-regulation of RAMP1 mRNA is unclear.
Direct interaction: Osteoblasts
In contrast to osteoclasts, osteoblasts are responsible for the deposition of bone matrix and bone formation (16). There is evidence of differential expression of calcitonin-family receptors in the primary cells and cell-lines of osteoblasts. RAMP2 mRNA is expressed in both populations, however, in a study of 16 samples of cultured human osteoblasts, the relative levels of expression of calcitonin receptors were assessed by polymerase chain reaction (PCR), with predominant expression of RAMP1 and CLR, and only small levels of RAMP2 (3). CTR mRNA was found only in the cell line and not osteoblasts (3,17).
In several in vitro experiments of CGRP and amylin, CGRP appeared to stimulate osteoblast proliferation, and conversely, CGRP receptor blockade inhibited proliferation (18). The concentration of CGRP peptide or receptor-antagonist required however, was 100-fold greater than that of amylin, suggesting a differential affinity for amylin over CGRP in this context (18). Separate antagonism of CGRP and amylin in these cell lines confirmed that there was a receptor subtype for CGRP distinct from amylin (19). The clinical significance for this is uncertain.
Indirect activity: Leptin and the sympathetic nervous system
As noted, there is increasing evidence of neural regulation of bone metabolism, which is well summarised elsewhere (9). Neural regulation in bone turnover was first described in a series of animal experiments, in which it was demonstrated that leptin deficient mice resulted in low sympathetic tone and high bone mass, an effect that was modulated by exogenous β-adrenergic agonism (20). It has been suggested that one pathway by which leptin influences bone turnover may be mediated by the sympathetic nervous system (9,21). This is supported by early studies that demonstrated that osteoblasts expressed β2-adrenergic receptors (20), and later, that beta agonism reduced, and antagonism increased bone mass in wild-type mice (22). Finally, a serotonin receptor knockout study provided evidence that leptin modulated serotonergic neurotransmission, and projected to ventromedial hypothalamic neurons, inhibiting bone mass accrual (23). Neuromedin U (NMU), a leptin regulated hypothalamic neuropeptide, has also been shown to have a role in bone formation (24).
CGRP similarly may modulate the sympathetic nervous system, as CGRP–/– mice have been found to have higher blood pressure and elevated sympathetic signals compared with wild-type mice (25,26). Furthermore, administration of leptin in a brain-injury model both promoted CGRP expression, and antagonism of CGRP partially negated the effects of leptin (27), and in a third study, inactivation of CGRP neurons negated the effect of leptin-induced anorexia (28). Finally, CGRP release from perivascular tissue was crucial for adjacent adipocyte release of leptin (29). Taken together, this raises the hypothesis of both a direct and indirect connection between CGRP and leptin mediated by the sympathetic nervous system. Leptin has multiple co-variates however, including gender, weight, age, dietary restriction and metabolic factors (30–32). Any study into the interaction between CGRP and leptin would be required to address these factors.
Knock-out studies
To further explore the role of CGRP on bone metabolism, several animal knock-out studies have been undertaken. In the first, CALCA, encoding calcitonin (CT) and CGRP, was studied (33). CT/CGRP–/– mice had significantly increased bone volume and bone formation at one and three months of age, and maintained bone mass after ovariectomy in contrast to wild-type mice who lost one-third of bone mass (33). Curiously, as stated previously, a further study of leptin receptor-deficient mice found an identical pattern of altered bone metabolism as the CT/CGRP knock-out mice (34). Finally, in a long-term study, α-CGRP–/– mice were found to have evidence of osteopenia, while CALCA–/– mice had evidence of both increased bone formation and resorption (35).
Commentary of the reading of CALCA–/– studies highlights the difficulty of the interpretation of these results due to the complex genotype of the animals, as well as the mixed genetic background of the mice (3).
Methodology
Study design
This is a prospective, longitudinal multi-centre cohort study conducted from June 2021 to July 2022 with participants actively recruited from two major tertiary hospital headache clinics (Alfred Health and Austin Health). The study is designed to investigate the effect of CGRP monoclonal antibody (mAb) administration on bone turnover. This study has received institutional review board approval (HREC 727/21, 22/Austin/08).
Research participants
Participants who met the ICHD-3 criteria (36) for chronic migraine and local regulations for the prescription of a CGRP monoclonal antibody were considered for inclusion in the study. Patients who at baseline had abnormal thyroid function, hypovitaminosis D, recent fracture, confounding medication (e.g. bisphosphonate) or bone turnover outside of the normal range at baseline were excluded from the study. In addition to standard care, participants underwent a fasting morning blood-test prior to administration of CGRP mAb and repeat fasting testing at a minimum of three-months following prescription. Clinical efficacy was determined by change in monthly headache days (MHD).
Markers of bone turnover
Under physiological conditions, bone resorption occurs in approximately ten days, and formation in three months (37). Markers of bone turnover allows for the monitoring of the homeostasis of bone formation and resorption (37). The International Osteoporosis Foundation (IOF) and the International Federation of Clinical Chemistry and Laboratory Medicine have recommended procollagen type I N-terminal propeptide (P1NP) and serum C-terminal telopeptide of type I collagen (CTX) as validated markers of bone formation and resorption, respectively (38).
P1NP is a reliable marker of bone formation as it has low variability, circadian variation and good assay precision (37,38). It is derived primarily from the proliferation of osteoblasts and fibroblasts (39). Carboxy terminal crosslinked telopeptides (CTX) is a degradation product of type 1 collagen of bone, and as such is not a direct marker of osteoclast activity. It is utilised as a marker of bone resorption, however is subject to significant variation with the circadian rhythm and post-prandial state (40,41). Thus, to be reliable, it should be obtained on a morning fasting sample.
Statistical analysis
The within-subject variability of bone turnover P1NP and CTX is 8% and 10% respectively (42). As such, the study was designed to detect a minimum of 10% difference within the group between the paired samples. Presuming a population variance of 225, 36 patients were required to provide 80% power with 95% confidence of finding a 10% difference.
Statistical analysis was performed using SPSS v28.0. Population characteristics were summarised with descriptive statistics. Longitudinal change was assessed with paired samples T-test for normally distributed, and Wilcoxon signed rank test for non-normally distributed data. Pearson correlation was used to assess correlation. Test results were considered significant when p < 0.05.
Results
A total of 54 patients were screened for the study, six patients were excluded for abnormal blood-tests at baseline, three patients were excluded due to a delay in collection of morning blood test of more than two hours, and 45 patients were included in the final analysis. The mean age of the cohort was 41.8 (SD 11.9), and 37/45 (82.2%) of the cohort was female. Population characteristics are summarised in Table 1. Reflecting local availability all patients were commenced on a mAb targeting the CGRP ligand; 41/45 (91.1%) galcanezumab and 4/45 (8.9%) fremanezumab.
Population characteristics.
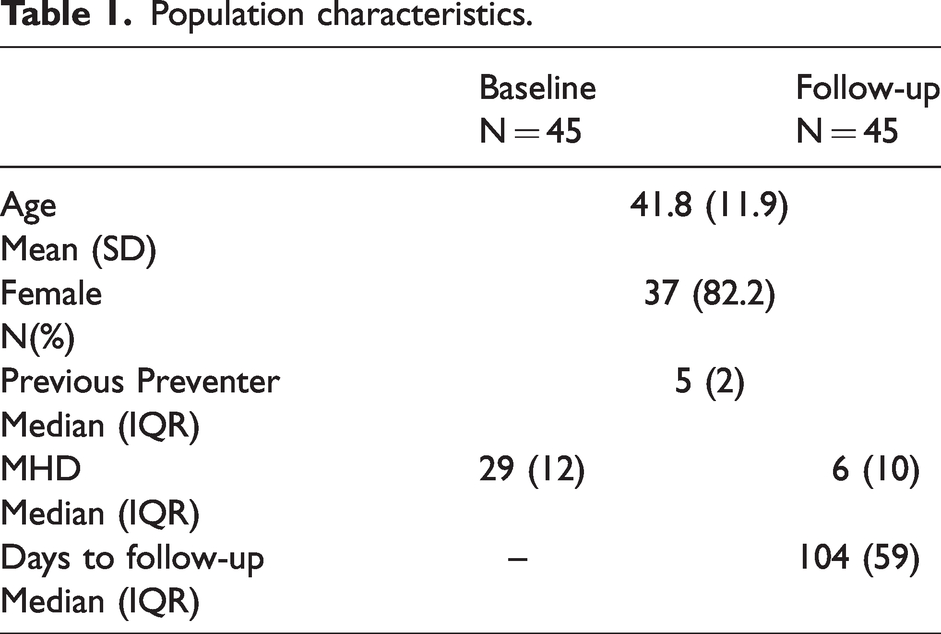
Bone-turnover markers at baseline and follow-up are presented in Table 2. Serum level of P1NP was significantly increased following CGRP mAb administration from 44.5 microg/L (IQR 22) to 51.5 (IQR 21) (z = −2.869, p = 0.004), with a median percentage change of 16.7% (IQR 37.9) (Figure 2). There was no significant change in CTX level. Repeat testing was planned to occur three months post commencement of CGRP mAb, but due to a variety of factors testing was delayed in some patients. The median time to follow up testing was 104 days (IQR 59). An exploratory analysis was undertaken to examine the effect of variation in time to follow-up and clinical efficacy on bone turnover. A Pearson correlation found no relationship between time of testing and percentage change in P1NP (r = 0.272, p = 0.074) or change in CTX (r = 0.011, p = 0.947). There was no correlation between percentage reduction in MHD and percentage change in CTX (r = 0.044, p = 0.785), or P1NP (r = −0.102, p = 0.510).
Change in bone turnover marker following administration of CGRP monoclonal antibody. P1NP; procollagen type I N-terminal propeptide, CTX; c-terminal telopeptide of type I collagen.

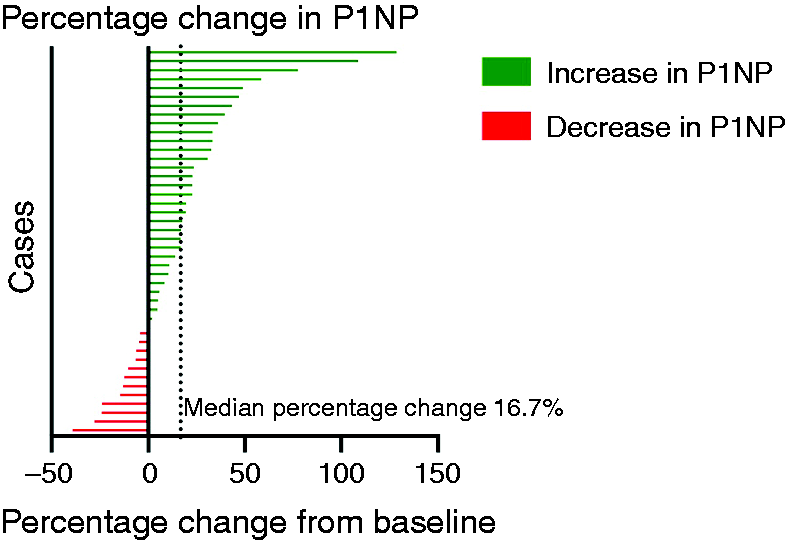
Percentage change in procollagen type I N-terminal propeptide (P1NP) after calcitonin gene-related peptide (CGRP) monoclonal antibody administration.
Discussion
This study is the first clinical study to investigate the effect of CGRP monoclonal antibodies on markers of bone turnover, and demonstrates a statistical increase in markers of bone formation following administration. Considering the pre-clinical evidence, it is significant that this study does not find any evidence that the use of CGRP monoclonal antibodies increases bone resorption. The study findings are concordant with the work of Hoff et al., who found evidence of increased bone formation following CALCA inhibition (33).
Interestingly, our study results have not reproduced the pre-clinical observations of CGRP activity on osteoblasts from which, we would presume reduced osteoblast proliferation and bone formation in a state of inhibition (18). This may be due to the relative action and affinity of a non-inhibited calcitonin-family peptide such as amylin (18), and/or the activation of an indirect mechanism resulting in net proliferation of osteoblast.
Leptin inhibition presents with an identical phenotype of bone formation, and as discussed, may have an inter-relationship with CGRP. Inhibition of CGRP may increase sympathetic nervous system signals (25,26), while increased sympathetic nervous system may inhibit leptin release (21). As discussed previously, a network (27,28), and direct (29) relationship between CGRP and leptin has been implied in several previous pre-clinical studies. Given the multiple co-variates of leptin (30–32), further investigation is required to explore this possible interaction.
Given the lack of evidence of bone resorption following administration of CGRP mAb, it appears that despite the pre-clinical evidence, this class of medications do not have a significant effect on osteoclast proliferation within three to four months of administration. One possible explanation is that under physiological conditions, including stimulation of M-CSF, RAMP1 mRNA levels are down-regulated, limiting the effect of CGRP and thereby CGRP inhibition on osteoclast activity (12). A second possible explanation is that inhibition of the CGRP ligand, as occurred with our patients, was less inciting than direct blockade of the receptor.
Other hypotheses for the lack of significant change in markers of osteoclast activity include the action of CGRP on osteoclasts may possibly occur ‘down-stream’ to counter-regulatory cross-talk, mitigating its effect (and thereby the effect of its inhibition) in this complex environment. Alternatively, there may be a secondary reactive change in osteoclast activity following the initial reported osteoblast biomarker, which was not captured due to study design and relatively short three-month follow-up period. Longer term study is required to investigate this possibility.
There are several limitations to this study. Firstly, this study was conducted during the COVID-19 pandemic and there was an unavoidable variability in the time of patient follow-up. While there was no correlation on Pearson analysis between time to follow-up and change in bone turnover marker, this is a possible confounder. Secondly, there are several sources of pre-analytical variability of bone turnover markers (38). Where possible, these have been controlled through the utilisation of paired samples and patient selection (i.e. patients without other comorbidity such as smoking, diabetes, thyroid disease, confounding medication etc.). Relative immobility or disturbed sleep due to uncontrolled disease prior to treatment was not possible to be controlled for. Immobility is associated with increased bone resorption however, which was not observed in our study, suggesting this is not a significant factor (38). Furthermore, no correlation was observed between percentage reduction in headache days and change in bone turnover markers, suggesting this was not a significant co-factor. Nevertheless, sleep and change in diet were not able to be assessed.
There are several further caveats to the generalisability of the study findings. This study population involved administration of a ligand-targeting CGRP mAb in a selected group of patients without other significant comorbidity, and may not be generalisable to CGRP-receptor targeted or small molecule CGRP antagonists. The study results, while reassuring, need to be replicated over a longer time-period. Finally, the relevance of our findings in states of increased bone-turnover such as post-menopause, acute fracture or increased calcium stress cannot be commented upon given all participants had normal bone turnover markers at baseline.
Conclusion
In otherwise homeostatic conditions, administration of a ligand-targeting CGRP monoclonal antibody is associated with a rise in markers of bone formation but not resorption. The clinical implications on fracture risk, while reassuring, are not certain. Further study is required to validate these findings over a longer time period in a larger cohort with different bone health states, and to examine the effect of CGRP monoclonal antibodies on bone density and explore any potential anabolic actions. Given the study findings, further investigation of the relationship between CGRP, and CGRP inhibition and leptin is also warranted.
Key findings
Short term use of CGRP monoclonal antibodies is associated with increased biomarkers associated with bone formation. Short term use of CGRP monoclonal antibodies have no effect on biomarkers associated with bone reabsorption.
Footnotes
Acknowledgments
The authors wish to sincerely thank Helmut Butzkueven for his guidance and the headache teams at the Alfred and Austin hospital. Figures created in biorender.com.
Contributions statement
The study was designed primarily by JCR with input from EJH and MSM. JCR and JB collected data. JCR performed primary analysis with extensive input from SSM, MSM and EJH. JCR wrote the main manuscript text with extensive input from all authors.
Data availability statement
The data in this study will be made available on application to the authors subject to local human ethics and research committee review and data sharing approval.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JCR has received funding from the Pharmaceutical Society of Australia and the Limbic supported by unrestricted educational grants from Viatris and Novartis respectively. MSM serves on the advisory board for Allergan, Novartis, Eli Lilly, Autonomic Technologies Inc and TEVA and has received payment for the development of educational presentations from Allergan, electroCore, Eli Lilly, Novartis and TEVA. EJH has served on advisory boards for Sanofi-Genzyme, Novartis, Teva, Eli Lilly, Allergan, Lundbeck, been involved in clinical trials sponsored by Novartis, Teva, Xalud, Cerecin, and has received payment for educational presentations from Allergan, Teva, Eli Lilly and Novartis SSM and JB report no potential conflict of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.