
Abstract
Background
Headache with neurologic deficits and cerebrospinal fluid lymphocytosis, previously also termed pseudomigraine with temporary neurologic symptoms and lymphocytic pleocytosis, is a self-limiting syndrome characterized by moderate to severe headache associated with focal neurological deficits occurring in the context of lymphocytosis in the cerebrospinal fluid. As a consequence of its rarity, data regarding headache with neurologic deficits and cerebrospinal fluid lymphocytosis is sparse. Therefore, we conducted this review to analyze data related to 93 patients of headache with neurologic deficits and cerebrospinal fluid lymphocytosis, to characterize their demographics, clinical manifestations, investigations and treatment options.
Methods
We performed a systematic review of cases reported through PubMed and Google scholar database, using Preferred Reporting Items for Systematic Reviews and Meta-Analyses protocol. Keywords used were ‘Headache with Neurologic Deficits and cerebrospinal fluid lymphocytosis’, ‘Headache with neurologic deficits and cerebrospinal fluid lymphocytosis syndrome’. The quality of the included studies was assessed using the Joanna Briggs Institute Critical Appraisal Tool.
Results
We analyzed a total of 93 cases of headache with neurologic deficits and cerebrospinal fluid lymphocytosis with a mean age of 28.8 years at onset. Seventy patients (75.2%) were adults, while 23 (24.7%) belonged to the pediatric age group. Comparing these groups, mean age at onset was 32.5 years and 14.3 years, respectively. The average duration of follow-up was 11.08 months. Thirty percent of patients experienced relapsing episodes of headache with neurologic deficits and cerebrospinal fluid lymphocytosis symptoms. The most common type of headache reported was unilateral severe throbbing episodic headache. Other associated symptoms included sensory deficit (60%) and motor deficits (54.8%). The least common symptoms were nystagmus and agraphia, which were reported in one patient each. Antiviral agents were a common treatment option in the acute phase (n = 23 patients [23.6%]), while Flunarizine was the most commonly used agent in the chronic setting (n = 3 patients [3.2%]). While most of the patients had normal brain magnetic resonance imaging, 20 patients had magnetic resonance imaging abnormalities, including (but not limited to) non-specific white matter lesions (eight patients) and meningeal enhancement (six patients). The most common electroencephalographic findings included diffuse and focal slowing. The mean cerebrospinal fluid opening-pressure was 240.5 mmH2O. Cerebrospinal fluid protein was elevated in 59 (63.4%) patients, with a mean value of 114 mg/dL. Two patients in our cohort were found to have cerebrospinal fluid oligoclonal bands.
Conclusion
Headache with neurologic deficits and cerebrospinal fluid lymphocytosis tends to affect young individuals with a slight male predominance. Unilateral severe throbbing episodic headache with associated hemi-paresthesia and hemiparesis were the most common symptoms based on our review. Elevated cerebrospinal fluid opening-pressure can be seen in headache with neurologic deficits and cerebrospinal fluid lymphocytosis syndrome. Early recognition of the syndrome is paramount. Antivirals were found to be among the most widely used treatments in the acute setting. Magnetic resonance imaging of the brain is mostly normal. Diffuse and focal slowing were among the most common electroencephalographic findings. Cerebral flow abnormalities on perfusion scans are not uncommon in headache with neurologic deficits and cerebrospinal fluid lymphocytosis. Prospective studies with a larger sample size are needed to validate our findings and guide the clinical care of these patients.
Introduction
Headache with Neurologic Deficits and cerebrospinal fluid (CSF) lymphocytosis (HaNDL) is a complex syndrome that is characterized by transient headaches and a wide array of transient neurological deficits, that are temporally associated with the onset of headache, and the presence of CSF pleocytosis. These episodic headaches, which are known to recur, can last for several hours and range from intermediate to severe intensity. CSF cultures are typically normal and lymphocyte-predominant CSF pleocytosis of >15 cells/µL is required for diagnosis (1) (Table 1). According to the International Classification of Headache Disorders 3rd edition (ICHD-3), the diagnostic criteria of HaNDL syndrome include episodes of migraine-like headache accompanied by or shortly preceded by onset of at least one of the following three, for greater than four hours: hemiparesthesia, dysphasia, hemiparesis, (2) along with CSF lymphocytic pleocytosis (>15 white cells per µl) and negative etiological studies. Additionally, there must be demonstration of development or worsening (alternately, improvement) of headache and transient neurological deficits in parallel to onset or worsening (alternately, improvement) of the CSF lymphocytic pleocytosis. Finally, the presentation is not better explained by another ICHD-3 diagnosis (2). Most cases of HaNDL syndrome appear in patients in their third or fourth decades of life, and the disease tends to preferentially affect males (3). Brain imaging is usually unremarkable, and the lack of other key indicators makes deducing the pathogenesis of the disease difficult.
The ICHD-3 diagnostic criteria of HaNDL syndrome.

The pathophysiology of HaNDL syndrome is not well understood, although many hypotheses exist. One such hypothesis suggests that infection and inflammation play a major role in the pathogenesis (4). Few cases of HaNDL syndrome in the literature have presented with viral prodromes, with seropositivity for agents such as Human herpesvirus (HHV)-7, Borrelia and Epstein-Barr virus (EBV) (4–6). Additionally, one of the brain magnetic resonance imaging (MRI) findings in patients with HaNDL syndrome includes leptomeningeal enhancement, a nonspecific finding that is occasionally associated with inflammatory and infectious states. In some pediatric cases, a viral prodrome precedes onset of HaNDL syndrome, further bolstering support for the infection-driven hypothesis. Another hypothesis that is frequently discussed is that of vasospasm and temporary reductions in brain perfusion as a potential contributor to the pathogenesis of HaNDL (7). This hypothesis arose when transcranial Doppler performed on patients with HaNDL syndrome revealed asymmetry and fluctuations in middle cerebral artery blood flow velocity. These asymmetries were found to be resolved when the HaNDL syndrome symptoms subsided (8). Transcranial dopplers and perfusion studies data of patients with HaNDL syndrome also revealed changes in cerebral blood flow with recovery over the following days, suggesting a role for hypoperfusion in the disease onset (9–12). In some of these patients, a cortical spreading depression pattern similar to that seen in migraine has been observed, suggesting that these vasospastic changes could induce a spreading depression that leads to the HaNDL syndrome presentation (13). Other mechanisms that have also been proposed include a potential role for autoimmunity. An immunohistochemical study of four HaNDL syndrome patients identified autoantibodies in two patients to CACNA1H, a subunit of T-type calcium channels (8). Clinical similarities between HaNDL syndrome and hemiplegic migraine (which is associated with a mutation in the same calcium channel complex) have led to speculation that autoimmunity may play a role in pathogenesis. Others have suggested a combination of hypotheses, such as an infectious/inflammatory etiology triggering autoantibody formation, and subsequent neurologic deficit in a spreading pattern (14). Regardless of the hypotheses, the ultimate pathophysiology is still unclear, and may include contributions from multiple causes.
Symptomatology of HaNDL syndrome includes headache, confusion, nausea, vomiting, visual disturbance, cranial nerve deficits, and sensorimotor disturbances. In addition to the headache in HaNDL syndrome, the most common associated symptoms are sensory symptoms followed by language disturbances and hemiparesis (15). The presenting symptom is often variable. When a patient presents with such symptoms, the possibility of HaNDL syndrome should be entertained. HaNDL syndrome is a diagnosis of exclusion. Inflammatory conditions in the differential diagnosis include encephalitis, neurosyphilis, meningitis, Lyme’s disease, and vasculitis (3). Besides these conditions, focal seizures and structural lesions, such as strokes or tumors, can also mimic symptoms of HaNDL syndrome, and must be ruled out.
The aim of this review is to compile and understand the various presentations, demographic attributes, current diagnosis and management of HaNDL syndrome. Our secondary outcomes are to evaluate the characteristics of electroencephalographic (EEG), MRI and CSF findings in HaNDL syndrome patients.
Methods
We performed a systematic review of cases reported on HaNDL syndrome. We followed the predesigned Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) protocol and PRISMA checklist 14 (online supplementary file 1) and standard for reporting the systematic review to the best of our capabilities (16).
Search strategy
A comprehensive search for case reports and case series on PubMed and Google Scholar database was conducted on 15 May 2022 by two independent investigators (MA and FA). The search included published case reports, case series, case illustrations, and letters reporting human cases, between January 1996 and June 2021, and was performed by using keywords: Headache with Neurologic Deficits and CSF lymphocytosis (HaNDL Syndrome), pseudomigraines.
Eligibility criteria
We performed a PubMed and Google Scholar search using the following terms: “HaNDL [title],” “Headache with Neurologic Deficits and cerebrospinal fluid Lymphocytosis,” “headache with neurologic deficits and cerebrospinal fluid pleocytosis,” “migraine with cerebrospinal fluid pleocytosis,” and “pseudomigraine with cerebrospinal fluid pleocytosis,” which was the original name used to describe the condition. The search yielded 57 studies, constituting 93 patients that met the ICHD-3 criteria for the diagnosis of HaNDL syndrome. Observational studies, review articles, letters to the editor and, and case series with duplication of patient data were excluded.
Selection of studies and data collection
Our systematic review was conducted following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines. PubMed and Web Sciences databases were systematically searched from January 1996 until June 2021, using the combination of the Boolean operators “OR” and “AND”, and the search terms Headache with Neurologic Deficits and CSF lymphocytosis (HaNDL Syndrome). Studies were exported to Endnote and duplicates were removed. We also performed a manual search for additional relevant studies using references of the included articles. The search was not limited to studies written in English.
By using this search strategy, a total of 150 articles were identified and screened. We excluded 50 articles which were not full-text, and outside of the January 1996–June 2021 publication range. The studies prior to 1996 had incomplete patient characteristics, including outcome, imaging and or CSF profile, making it difficult to include in our review. Two investigators (MA-C and FA) then independently reviewed 100 articles, including abstracts and full manuscripts, and selected the articles based on the ICHD-3 diagnostic criteria of HaNDL Syndrome (Table 1). Any disagreement was reviewed by a third investigator (KG) and disagreement was resolved by consensus. Forty-three articles were further excluded, as they had incomplete information on demographics or were difficult to comprehend. This left us with a total of 57 case reports and case series (Figure 1) (1,5–7,10,13,17–68).
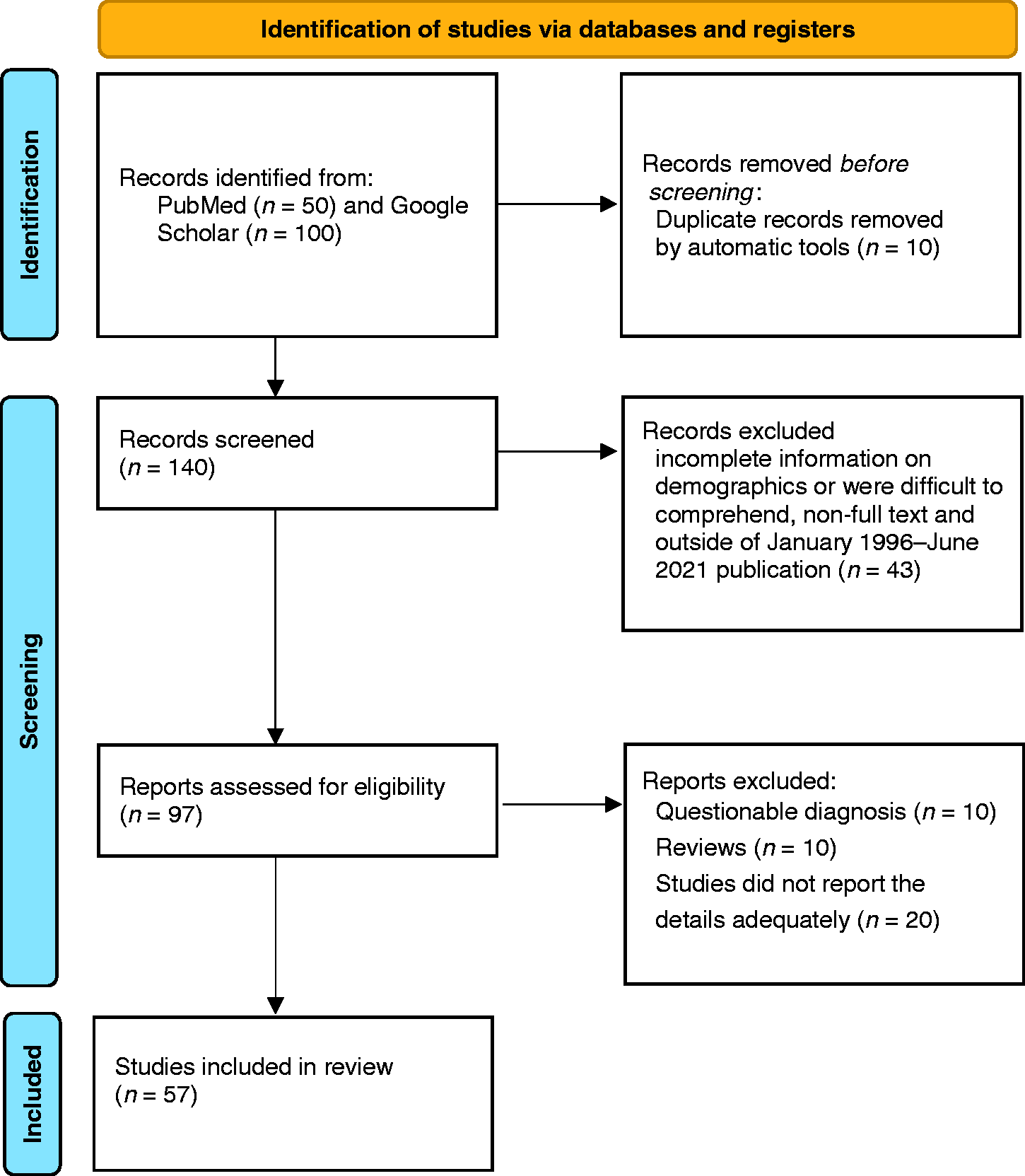
PRISMA flow diagram for the selection of studies.
All data were summarized descriptively, including country of reporting, age, sex, duration of headache and other neurological deficits, characteristics of headache, concomitant symptoms, imaging and electroencephalogram characteristics, comorbidities and therapeutic interventions. Pediatric age was defined as younger than 18 years, and adult as 18 years and older.
Quality assessment
We assessed the quality of the included studies (case reports and case series) using the Joanna Briggs Institute (JBI) Critical Appraisal Tool. Two authors (FA and ND) independently assessed each study for risk of bias. Discrepancies were resolved by a third reviewer (YE).
Outcomes
The primary outcome of our systematic review of cases is to evaluate the demographic characteristics, presenting symptomatology, variation in presentation of HaNDL syndrome, and treatment interventions in both the acute and chronic settings. The secondary outcome is to evaluate the CSF profile, EEG and MRI characteristics of HaNDL syndrome patients.
Results
We analyzed a total of 57 published case reports and series, constituting a total of 93 patients, which fulfilled the inclusion criteria for this review.
Demographics, comorbidities, outcomes and follow-up duration
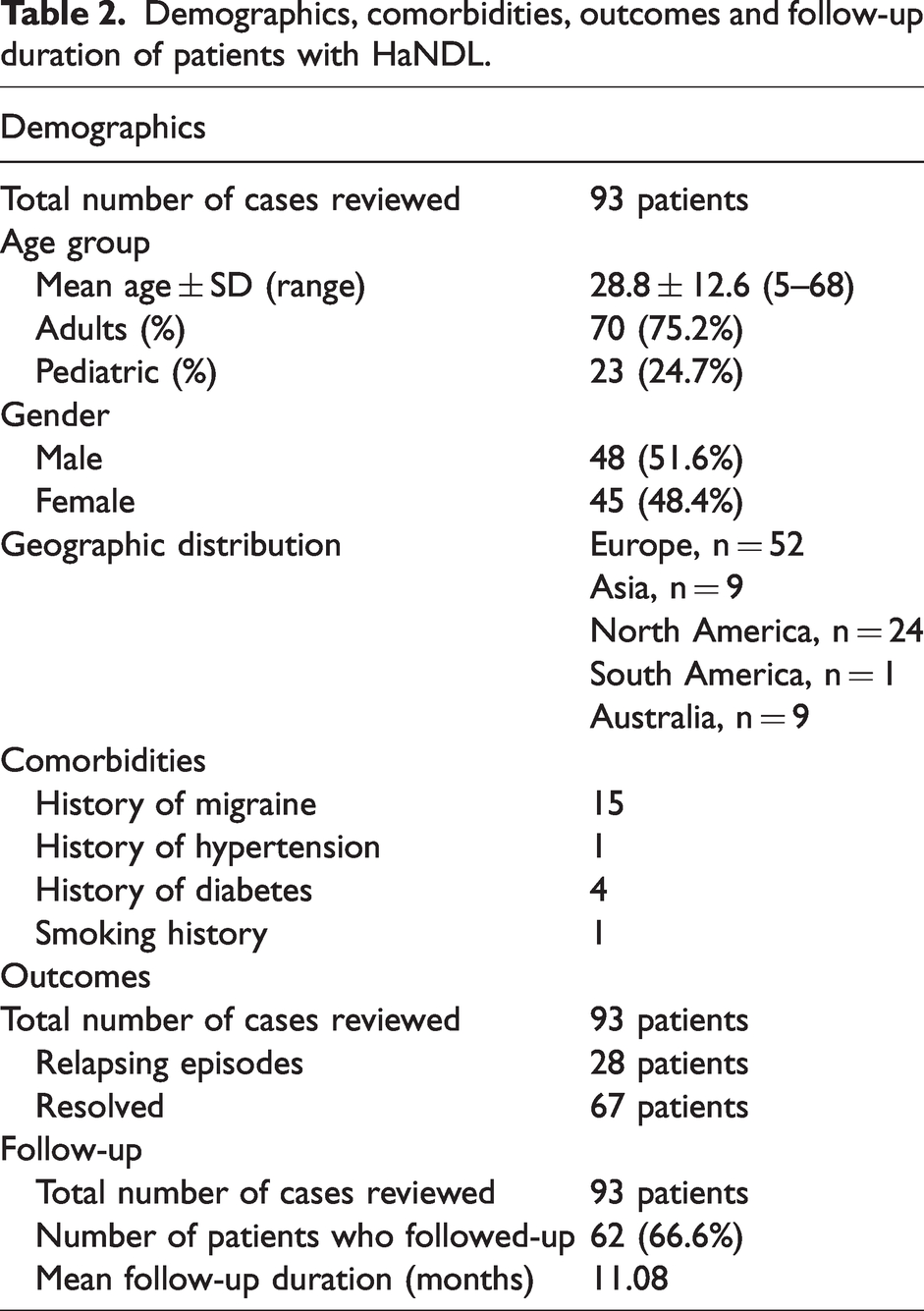
Of the 93 cases reviewed, 70 patients (75.2%) were adults while 23 patients (24.7%) were of pediatric age. The sample was comprised of 48 males (51.6%) and 45 females (48.4%). The mean age of onset was 14.3 years for pediatric patients, and 32.6 years for adult patient. Analyzing the associated comorbidities in these patients, 15 patients already carried a diagnosis of migraine headaches, four patients had diabetes mellitus, and one patient had hypertension. Out of the 93 cases reviewed only one patient reported smoking. The mean follow-up duration was 11 months. Twenty-eight patients had a relapsing course. Most of the patients were from Europe (55.9%), followed by North America (25.8%), Asia (9.7%), Australia (7.5%) and South America (1.1%), respectively (Table 2).
Demographics, comorbidities, outcomes and follow-up duration of patients with HaNDL.
Presenting symptomatology
Migraine-like headache was present in all 93 patients (100%). Headache was further classified into episodic headache 79 (84.9%), and single headache (4.3%), of which thunderclap headache was reported in one case (55). We defined headaches re-occurring within three months as episodic based on ICDH-3 description of HaNDL timeframe (69). Out of the 30 cases that reported severity, 24 had severe, four moderate, and two mild headaches. Of the 42 cases that reported headache laterality, 35 patients had left-sided, four right-sided, and three bilateral headaches. Headache character was described in 38 patients (41%), and was throbbing in most cases. Nausea and vomiting were reported in association with headache in 47 patients (50.5%). Sensory symptoms were reported in 56 patients (60%), whereas motor symptoms were reported in 51 patients (54.8%). Unilateral paresthesia sparing face was the most common sensory complaint (37 patients), whereas hemiparesis was the most common motor deficit (45 patients). Aphasia (mostly expressive) was reported in 44 patients (47.3%), and dysarthria was reported in 10 patients (10.7%). Confusion was reported in 31 patients (33.3%). Papilledema was reported in 15 patients (16.1%), abducens nerve palsy in four patients (4.3%), diplopia in three patients (3.2%), and nystagmus in one patient. Ataxia was reported in six patients (6.4%) (Table 3).
Clinical characteristics of patients with HaNDL.

Imaging and EEG findings
The most common brain imaging performed for patients presenting with HaNDL syndrome symptoms was MRI, which was found to be abnormal in 20 patients (25.6%) out of the reported MRI results in 78 patients. In the majority of the patients imaging did not reveal abnormalities. The reported abnormal findings included nonspecific white matter lesions in eight patients, meningeal enhancement in six, lesions of corpus callosum in four, diffusion restriction in two and hydrocephalus in one patient (Table 4). It is worth noting that two patients whose brain MRIs demonstrated white matter lesions had a history of migraine while the other six patients did not have a history of migraine or other comorbidities such as, diabetes or hypertension or smoking history. Migraine and cardiovascular risk factors have been associated with white matter lesions on MRI (70,71). MRI has a utility in aiding in the diagnosis of HaNDL syndrome, as Segura et al. (10) describe a case where multimodal MRI determined that a suspected stroke in a patient was truly a stroke mimic. Imaging revealed the absence of an infarction zone, no alterations on diffusion weighted imaging (DWI), and an extensively delayed perfusion in the let hemisphere. MR-angiography then revealed reduced left middle cerebral artery (MCA) diameter relative to the right MCA, suggesting a perfusion/diffusion mismatch that ultimately led to further workup and diagnosis of HaNDL syndrome. In doing so, the need for IV thrombolysis was eliminated.
Diagnostic Test Results of Patients with HaNDL.

MRI: magnetic resonance imaging, EEG: Electroencephalogram, CT: computed tomography, CTP: Computer tomography profusion, MTT: mean transit time, TTP: time to peak, MCA: middle cerebral artery, SPECT: single-photon emission computerized tomography.
Transcranial Doppler Ultrasonography or Computed Tomography (CT) perfusion or single-photon emission computerized tomography (SPECT) studies were abnormal in 11 patients. CT perfusion studies were obtained in six cases. The reports showed increased MCA velocities contralateral to the symptoms in two patients, decreased contralateral MCA velocity in one patient and asymmetrical MCA velocities in one patient. CT perfusion imaging revealed hypoperfusion in the temporo-parieto-occipital region in three patients and the frontal region in two patients. One patient showed increased mean transit time (MTT) and time to peak (TTP) with normal cerebral blood flow (CBF) in the left hemisphere (Table 4). One patient demonstrated decreased blood flow in the temporal area on SPECT scan. Diagnostic cerebral angiogram findings were reported in 25 cases in which 18 had normal angiogram findings with exclusion of vasospasm, arterial dissection, and venous sinus thrombosis. One case was found to have incidental chronic arterial on cerebral angiogram. Additionally, there were two cases in which angiogram may have precipitated a HaNDL syndrome event (59).
EEG findings were reported for 65 cases, out of which 37 (56.9%) were abnormal and 28 (43.1%) were normal. The patterns of slowing seen were generalized in 20 patients, and focal in 17 patients. The findings of focal slowing were mostly seen in frontal and temporal regions. (Table 4). Recurrence of clinical symptoms were found to correlate with EEG findings, specifically asymmetric slowing (5). Despite this correlation, the pathophysiology behind these EEG changes is not clearly explained yet. Focal hypoperfusion and intracranial vasomotor changes were proposed after some studies correlated areas of focal hypoperfusion to the same-region focal slowing on EEG (22,50).
CSF characteristics
The diagnosis of HaNDL syndrome in all patients was made according to the ICHD-3 criteria which included performing lumbar puncture showing lymphocytic pleocytosis and negative etiological studies. However, the specific result values were reported in 77 (82.7%) cases only. Cerebrospinal Fluid (CSF) opening pressure was reported in 34 (36.5%) patients, with a mean value of 240.5 mmH2O. CSF lymphocytes were reported in 77 (82.7%) patients, with a mean value of 147.5/mm3. CSF protein was reported in 59 (63.4%) patients, with a mean value of 114 mg/dL. Two patients in our cohort were found to have CSF oligoclonal bands (Table 5). Interestingly, one of the cases meeting all the diagnostic criteria for HaNDL syndrome was evaluated further and resulted a positive CSF PCR test for HHV-7. Notably, the case showed raised serum HHV-7 IgG titers, suggesting a recent infection, which possibly explains how the criteria for HaNDL syndrome were fulfilled. Authors of that case report suggest a possible correlation of HaNDL syndrome with an immune-mediated reaction after a viral infection (5). Follow-up CSF analysis was reported in 23 cases, with universal reduction of lymphocytic pleocytosis associated with resolution of transient neurologic deficits. The degree of reduction varied significantly, as did the time period over which the reduction occurred (1,3,17–20,23,24,26,29,31,34,35,43,47–49,53,56,60–62,65).
CSF analysis in patients with HaNDL.

Serum antibodies
A variety of serum antibodies were found in a limited number of patients. These include antibodies to Epstein–Barr virus (EBV), Cytomegalovirus (CMV), Varicella zoster virus (VZV), Coxsackievirus, as well as anti-Thyroid peroxidase antibodies (TPO) antibodies. Serology was positive in one patient for lgG antibodies against Coxsackie virus and VZV, and CSF work-up for anti-viral IgM/G antibodies was negative. Five patients (5.3%) tested positive for antibodies to three DNA repair proteins in the serum. These proteins include mitogen-activated protein kinase-4, DNA-dependent protein kinase catalytic subunit, and DNA excision repair protein ERCC-6.
Treatment options in the acute and chronic phases
As the mainstay treatment of HaNDL syndrome is symptomatic, our review demonstrated that a wide variety of treatment strategies were used in managing the syndrome in both the acute and chronic settings. Majority of these treatment options were employed in the acute setting. Regarding the acute treatment of HaNDL syndrome, antiviral agents (acyclovir and ganciclovir) were the most commonly used (n = 23 patients [23.6%]), followed by antibiotics in 14 patients (15%) and therapeutic lumbar puncture (13%). The use of antibiotics is easily explained by the fact that meningitis is one of the most common conditions on the differential diagnosis of HaNDL syndrome. Other treatment options that were reported in the acute settings include intravenous steroids in 11 patients (11.8%), analgesics in 11 patients (11.8%), acetazolamide in 10 patients (10.7%), intravenous thrombolytics in six patients (6.4%), Non-steroidal anti-inflammatory agents in seven patients (7.5%), antiemetics in five patients (5.3%), antihypertensives in four patients (4.3%), and intravenous hydration in four patients (4.3%) (Table 6). Less commonly used treatments in the acute setting included benzodiazepines, magnesium, anticonvulsants, heparin, sumatriptan and neuroleptics. The average duration for the acute treatment was 6.27 days. In the chronic settings, on the other hand, Flunarizine was the most commonly used agent (n = 3 patients [3.2%]), followed by Acetazolamide in two patients (2.1%) and corticosteroids in two patients (2.1%). Other less commonly used options in the chronic setting included NSAIDS, Metoclopramide, Acetaminophen and Verapamil (Table 7).
Acute treatment in patients with HaNDL.

IV: Intravenous; NSAIDS: Nonsteroidal anti-inflammatory drugs; IV-tPA: Intravenous tissue-plasminogen activator.
Chronic treatment in patients with HaNDL.

Discussion
Transient Headache and Neurologic Deficits with Cerebrospinal Fluid Lymphocytosis (HaNDL syndrome) is a headache disorder in which patients experience intermittent severe to moderate headache attacks with focal neurologic deficits with distinct cerebrospinal fluid findings (lymphocytic pleocytosis) (72). This syndrome is a rare mimicker of both stroke and migraine with aura, making its correct identification extremely important (7).
Various potential causes of HaNDL syndrome have been discussed. Inflammatory causes, infectious/viral etiologies, and spreading depression triggered by transient hypoperfusion have all been postulated as the possible underlying pathophysiology. Leptomeningeal enhancement on imaging has been described as an indicator of elevated intracranial pressure (ICP) (7), which could contribute to the presentation. The viral prodrome that is often seen prior to symptom onset also merits consideration. Finally, Doppler studies that demonstrate hypoperfusion in patients presenting with HaNDL syndrome adds further support for the vasospastic mechanism of disease (41). The pathophysiology of HaNDL syndrome is complex, and further investigation is needed to better elucidate the underlying mechanisms.
Our study reviews 93 patients with diagnosed HaNDL. According to the National Institute of Health, this condition is usually diagnosed in adulthood (65), although age range of seven to 52 years has been reported in individuals with this condition. According to our review the mean age of presentation, in years, was 28.8 ± 12.6. Although the condition is diagnosed mainly in adults, 24.7% of the studied patient population were in pediatric age range. Demographic information was further analyzed based on gender, which showed a very slight male predominance (51.6% male versus 48.4% female). This finding aligns with the assertion that HaNDL is more frequent in men (1). Further demographics in our analysis included the continent of origin for each case report/series. Europe contributed the largest studied patient population (55.9% of all), followed respectively by North America (25.8%), Asia (9.7%), Australia (7.5%), and South America (1.1%).
The presence of sensory symptoms (n = 56), motor symptoms (n = 51) and aphasia (n = 44) demonstrate that the current ICHD-3 criterion B1 appropriately reflects the presenting symptomatology of transient neurologic defects in these patients. Additionally, an elevated mean CSF lymphocyte count of 147.5 cells/mm3 was reported in 77 patients. The number of cases with viral CSF positivity (n = 5) and malignancy (n = 0) do not account for this generalized elevation. These findings suggest that in HaNDL syndrome, lymphocytic pleocytosis frequently presents with negative etiological testing, corroborating the ICHD-3 criterion B2. CSF lymphocytosis was discovered after initial presentation of transient neurologic deficits, supporting existing criterion C1 regarding discovery of lymphocytic pleocytosis. Many patients continued to have clinically defined lymphocytic pleocytosis for weeks to months after the initial episode with no symptoms. There was no observed pattern between treatment modality and speed of resolution of lymphocytic pleocytosis. Interestingly, one case presented with normal CSF analysis on admission, but developed lymphocytic pleocytosis six days later (62). These findings suggest that while a parallel temporal relation exists between lymphocytic pleocytosis and the presence of transient neurologic deficits, it may not be perfectly aligned. A degree of pleocytosis may persist after resolution of symptoms, and pleocytosis may also develop after neurologic deficits have been present. Criterion C2 is therefore generally appropriate, but it may not fully capture this slight temporal variability or the degree of variability in pleocytosis reduction.
Moreover, our data offers further support that an abnormal EEG pattern, predominantly diffuse slowing, is observed in HaNDL syndrome, as it was present in 46.2% of cases in our study. This is in line with previous literature describing intermittent slowing in delta or theta range on EEG as suggestive of HaNDL diagnosis (1). However, diffuse or focal slowing on EEG is not a specific finding, and can been seen with other primary headache disorders such as migraine (73–75). Our study found abnormal MRI findings in 20 (25.6%) out of the 78 patients in which MRI was performed, with the most prominent findings being non-specific white matter lesions (10%), meningeal enhancement (7.5%) and corpus callosum lesions (5.1%). This is in contrast with findings by Gómez-Aranda et al. that reports mostly normal MRI findings with the exception of one patient (9). There are previous studies that suggest that abnormal MRI findings may be present in some cases of HaNDL syndrome, including a case of gray matter swelling (37), CSF enhancement in temporal and occipital sulci, and corresponding hypoperfusion that mimic the abnormal MRI findings of hemiplegic migraine (37). This suggests potential variability and complexity in the diagnostic role of MRI in HaNDL syndrome.
One of the strengths of the study design was detailed study of individual cases, which allowed us to obtain detailed information regarding disease course, symptoms, treatment options, and ultimate outcomes for each patient. To our knowledge, this is the largest systematic review on HaNDL syndrome, including both pediatric and adult patient populations. This study comprehensively outlined the symptom prevalence as well as the current treatment practices in the acute and chronic setting. Additionally, we reviewed the common imaging, CSF and EEG findings in HaNDL syndrome patients. One limitation of our study, however, is the sample size. Our study addresses 93 cases with HaNDL syndrome, while excluding studies where individual patient data was not available. Although this was done to improve the quality of findings, it does reduce the final sample size. Despite this limitation, our study does address a relatively sizeable number of cases in the context of existing literature, and we strongly believe it will contribute to furthering a better understanding of HaNDL syndrome.
In conclusion, HaNDL syndrome is a rare syndrome with poorly understood pathophysiology that can present with a wide variety of neurological symptoms. This is the largest systematic review on this topic that aims to compile many cases to help examine the commonality of the presenting symptoms, treatment options, imaging, CSF and EEG profiles. Neurologists should maintain a high index of suspicion for HaNDL syndrome when encountering headache with neurological deficits. Prospective studies with a larger sample size are needed to validate our findings and guide the clinical care of patients with HaNDL syndrome.
Article Highlights
Headache with Neurologic Deficits and cerebrospinal fluid (CSF) lymphocytosis (HaNDL) is a rare headache syndrome that tend young individuals with a slight male predominance Early recognition of the syndrome is critical as our review showed that antiviral agents were the most common treatment in the acute setting Elevated CSF opening pressure can be seen in HanDL syndrome Brain MRI was normal in the majority of HaNDL cases EEG findings usually include focal slowing which are non-specific
Supplemental Material
sj-pdf-1-cep-10.1177_03331024231157694 - Supplemental material for Transient headache and neurological deficits with cerebrospinal fluid lymphocytosis syndrome: A comprehensive systematic review of 93 patients from 57 studies
Supplemental material, sj-pdf-1-cep-10.1177_03331024231157694 for Transient headache and neurological deficits with cerebrospinal fluid lymphocytosis syndrome: A comprehensive systematic review of 93 patients from 57 studies by Mustafa Al-Chalabi, Prajwal Hegde, Fahham Asghar, Nameer Aladamat, Nicholas Delcimmuto, Khaled Gharaibeh, Mohammad Samara, Yasar Esengul, Naeem Mahfooz and Ajaz Sheikh in Cephalalgia
Footnotes
Author contributions
Conceptualization: FA, MA-C, PH, ND, KG, NA, MS. Data Curation: KG, Investigation: YE and MA-C. Original Draft: AS, NM. Review and Editing: MA-C.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.