
Abstract
Background
Greater occipital nerve blockade for the prevention of chronic migraine has a limited evidence base. A robust randomized double-blind, placebo-controlled trial is needed.
Methods
This double-blind, placebo-controlled, parallel-group trial, following a baseline period of four weeks, randomly assigned patients of chronic migraine 1:1 to receive four-weekly bilateral greater occipital nerve blockade with either 2 ml of 2% (40 mg) lidocaine (active group) or 2 ml of 0.9% saline (placebo) injections for 12 weeks. The primary and key secondary efficacy endpoints were a change from the baseline in the mean number of headache and migraine days and the achievement of ≥50% reduction in headache days from baseline across the weeks 9–12 respectively. Safety evaluations included the documentation and reporting of serious and other adverse events.
Results
Twenty-two patients each were randomly allocated to the active and placebo group. Baseline demography and clinical characteristics were similar between the two groups. Mean headache and migraine days at baseline (±SD) were 23.4 ± 4.4 and 15.6 ± 5.7 days in the active group and 22.6 ± 5.0 and 14.6 ± 4.6 days in the placebo group respectively. The active group compared to the placebo had a significantly greater least-squares mean reduction in the number of headache and migraine days (−4.2 days [95% CI: −7.5 to −0.8; p = 0.018] and −4.7 days [95%CI: −7.7 to −1.7; p = 0.003] respectively). 40.9% of patients in the active group achieved ≥50% reduction in headache days as compared with 9.1% of patients receiving a placebo (p = 0.024). Overall, 64 mild and transient adverse events were reported by 16 patients in the active group and 15 in the placebo. No death or serious adverse events were reported.
Conclusion
Four-weekly greater occipital nerve blockade with 2% lidocaine for 12 weeks was superior to placebo in decreasing the average number of headache and migraine days in patients with chronic migraine with a good tolerability profile.
Clinical trial.gov no. CTRI 2020/07/026709
Introduction
Chronic migraine (CM) with an estimated worldwide prevalence of 2% is defined as headaches occurring on ≥15 days/month for more than three months, having features of migraine headache on at least eight days/month (1,2). CM is a highly disabling disorder (3,4) that imposes substantial direct and indirect economic costs (5) and often co-exists with acute migraine medication overuse which makes the treatment challenging (6,7). Currently, Onabotulinum toxin A (OBT-A) and monoclonal antibodies targeting the calcitonin-gene-related peptide receptor, or its ligand, are the only approved treatments for the prevention of CM (7,8). However, both these treatments are costly and hence there is a need to develop low-cost treatment options for the prevention of CM.
Greater occipital nerve blockade (GONB) is postulated to modulate the pain pathway through its connections at trigemino-cervical complex (TCC) (9,10). At TCC, there is a convergence between the trigeminal and upper cervical sensory afferents (greater occipital nerve fibers are predominantly derived from C2) which in turn is connected to higher pain-modulating structures in the brainstem and rostral pain pathways (11–13). GONB is easy to administer, safe, and offers a low-cost treatment to CM patients (14). Cuadrado et al. (15) assessed the short-term clinical efficacy of GONB in chronic migraine patients and found it to be significantly superior to placebo in reducing the number of moderate to severe or any headache during the week following the injection. However, its effectiveness for the long-term prevention of CM lacks a good evidence base (16). Three previous placebo-controlled double-blind randomized trials of GONB in CM had methodological limitations and produced conflicting results (17–19). Given the limited evidence of GONB for the long-term prevention of CM, we conducted a 12-week double-blind placebo-controlled trial to evaluate the efficacy, safety, and side effect profile of four-weekly bilateral GONB for the preventive treatment of CM using the norms laid down by the International Headache Society (IHS).
Methods
Trial oversight
The final study protocol, informed patient consent forms, and accompanying materials were reviewed and approved by the institutional ethics committee of Maulana Azad Medical College and associated G B Pant Institute of Post Graduate Medical Education and Research (GIPMER), New Delhi, India. The study was conducted as per the International Council for Harmonization Good Clinical Practice regulations and guidelines. The study follows the tenants of the Declaration of Helsinki. The authors take full responsibility for the conduct of the trial, adherence to the protocol, the accuracy and completeness of the data and analyses, and the reporting of adverse events. Informed written consent for participation in the study was taken from all participants at the start of the screening (Clinical trial registry of India CTRI 2020/07/026709).
Trial participants
Patients were recruited from Headache Clinic at GIPMER, New Delhi, India from 29 July 2020 through 27 April 2021. Inclusion criteria were age ≥18 years to ≤65 years, and fulfilment of the criteria of CM as per International Classification of Headache Disorders, third version (ICHD-3) (headache of any duration and severity for ≥15 days/month and headache meeting ICHD-3 criteria for migraine for at least eight days/month during the four weeks of baseline period) (2). Exclusion criteria were use of migraine preventive drugs during the three months prior to enrolment, pregnant women, patients with known allergies to lidocaine, and patients with a history of dementia, psychosis, and severe depression.
Trial design
This randomized, double-blind, placebo-controlled, parallel-group trial consisted of a baseline period of four weeks followed by a double-blind treatment phase (DBTP) for 12 weeks and then an open-label treatment phase for another 12 weeks (Figure 1). This paper describes the results of DBTP only. The patients were screened (week −4 to week 0) using a structured proforma by a neurology resident (AT) prior to their enrolment in the trial. Those not eligible were excluded (Figure 1). Randomization was done by a computer-based block randomization chart and patients were allocated to groups A or B (1:1) by another neurology resident (VD). Concealment was done using the opaque sealed envelope method. VD prepared injections for GONB. Both the injections contained colorless 2 ml of clear liquid in a 5 ml syringe with 26 number needle and were indistinguishable from each other. The consultant neurologist (DC), who was unaware of the content of the injections, applied GONB as per the standardized procedure (14). Briefly, the following procedure was followed: the site of injection was marked i.e., approximately two-thirds of the distance on a line drawn from the center of the mastoid to the external occipital protuberance only if these points exhibited conspicuous pain sensitivity to pressure. Prior to giving the injection, on the marked point and an area surrounding it with a 3 cm diameter, lignocaine jelly was applied to mask the effect of numbness following the GONB by lidocaine. GONB was given 10 minutes after ascertaining that the area was numb using a superficial pinprick. Those who did not achieve adequate local anesthesia were given lidocaine gel application again and the numbness was ascertained after another 10 minutes. 2 ml of 2% (40 mg) lidocaine and 2 ml of 0.9% saline were injected bilaterally in patients of group A and B respectively. Patients were observed for 30 minutes post-injection for any treatment-emergent adverse effects (TEAE). There was a total of four visits (0 weeks to 12 weeks) during the DBTP. AT who was unaware of the trial group assignment, documented the outcome including treatment-emergent adverse events (TEAE).

Trial design.
Trial endpoints
The primary efficacy endpoint was change in the mean number of headache days from the baseline period (week −4 to week 0) to the last four weeks of the DBTP (weeks 9–12). Key secondary endpoints were change in the mean number of migraine days and the achievement of at least a 50% reduction in the mean number of headache days during the same time. Other secondary endpoints were a change during the same time in mean cumulative headache hours (CHH), mean visual analog scale (VAS) score for attack severity (scored from 0–10), mean acute migraine treatment (AMT) days, and mean 6-item Headache Impact Test (HIT-6) score (higher score indicating greater disability) (20). Secondary endpoints that were measured at baseline and at the end of 12 weeks (weeks 0–12) included changes in mean Migraine Disability Assessment Score (MIDAS) (a five-item self-administered questionnaire with a higher score indicating greater disability) (21), a 3-dimension Migraine-Specific Quality-of-life Questionnaire (MSQ) score (higher score indicating better quality of life) (22), and Clinical Global Impression Severity (CGI-S) score that rates the clinician’s view of the patient’s global functioning (higher score indicating greater severity) (23). Safety evaluations during DBTP included the documentation and reporting of TEAE including serious adverse events (SAE) using a structured proforma in all randomized patients who received at least one dose of the trial regimen.
Statistical analysis
Estimates based on a previous trial (19) comparing GONB with placebo in CM patients predicted that a total of 30 patients completing the trial would provide 80% power to detect a mean ± SD difference of 6.6 ± 6.4 in mean number of headache days per month between the active group and placebo at two-sided alpha level of 0.05. Keeping a 20% loss to follow-up, a minimum of 36 (18 in each group) patients were needed for the study. Efficacy analysis using modified intention to treat included all patients who received at least one injection of GONB during DBTP and had at least one four-weekly follow-up data. For the primary and secondary endpoints that were measured at three time points (four, eight, and 12 weeks), the least-squares mean (LSM) at each time point was calculated with a linear mixed-effects model that included participants as a random effect, and the number of headache days during the four-week observation period and years since migraine onset as covariates. The model included fixed effects for the treatment group; medication overuse (presence vs absence); study month in the DBTP (month 1 was weeks 1–4, month 2 was weeks 5–8, and month 3 was weeks 9–12); and the month-by-treatment group interaction without any imputation for missing data. We reported the LSM change from baseline for each treatment group, treatment difference compared with placebo, 95% CI, and p values for pairwise comparison. The secondary endpoints that were measured at only baseline and at the end of 12 weeks, such as changes in mean MIDAS score, MSQ score, and CGI-S score, were analyzed using a linear model with treatment group and medication overuse (presence vs absence) as factors and baseline scores and years since migraine onset as covariates. The last observation carried forward (LOCF) method was used for imputing missing data for these secondary efficacy endpoints. For the 50% responder secondary endpoint, we used a stratified Cochran-Mantel- Haenszel test and LOCF for imputing missing data. We reported adjusted odds ratios (OR) compared with placebo, 95% CI, and p values. The level of significance was set at p < 0.05.
To control the type I statistical error rate at 0.05, a hierarchical testing procedure was applied, with a pre-specified sequence of comparisons beginning with the primary endpoint and proceeding to two key secondary endpoints in the order given in the protocol. Each comparison was performed only if the preceding comparison had a two-sided p-value of less than 0.05. Results are presented in the sequence in which endpoints were evaluated. If these comparisons were found to be significant, all other secondary endpoints were compared irrespective of a statistically significant p-value (less than 0.05). Statistical analysis was performed using R statistical software (v 4.1.2; R core Team 2021).
Results
Patients
A total of 44 patients were randomly assigned to one of the two groups, 22 to the active group and 22 to the placebo. Forty-two patients completed the DBTP and one patient in each group discontinued treatment after receiving two doses due to COVID-19 related lockdown. Thus all 44 patients were available for efficacy analysis (Figure 2). Baseline demographics and clinical characteristics were similar between the two groups. Mean four-weekly headache and migraine days at baseline (±SD) were 23.4 ± 4.4 and 15.6 ± 5.7 days in the active group and 22.6 ± 5.0 and 14.6 ± 4.6 days in the placebo group respectively (Table 1).
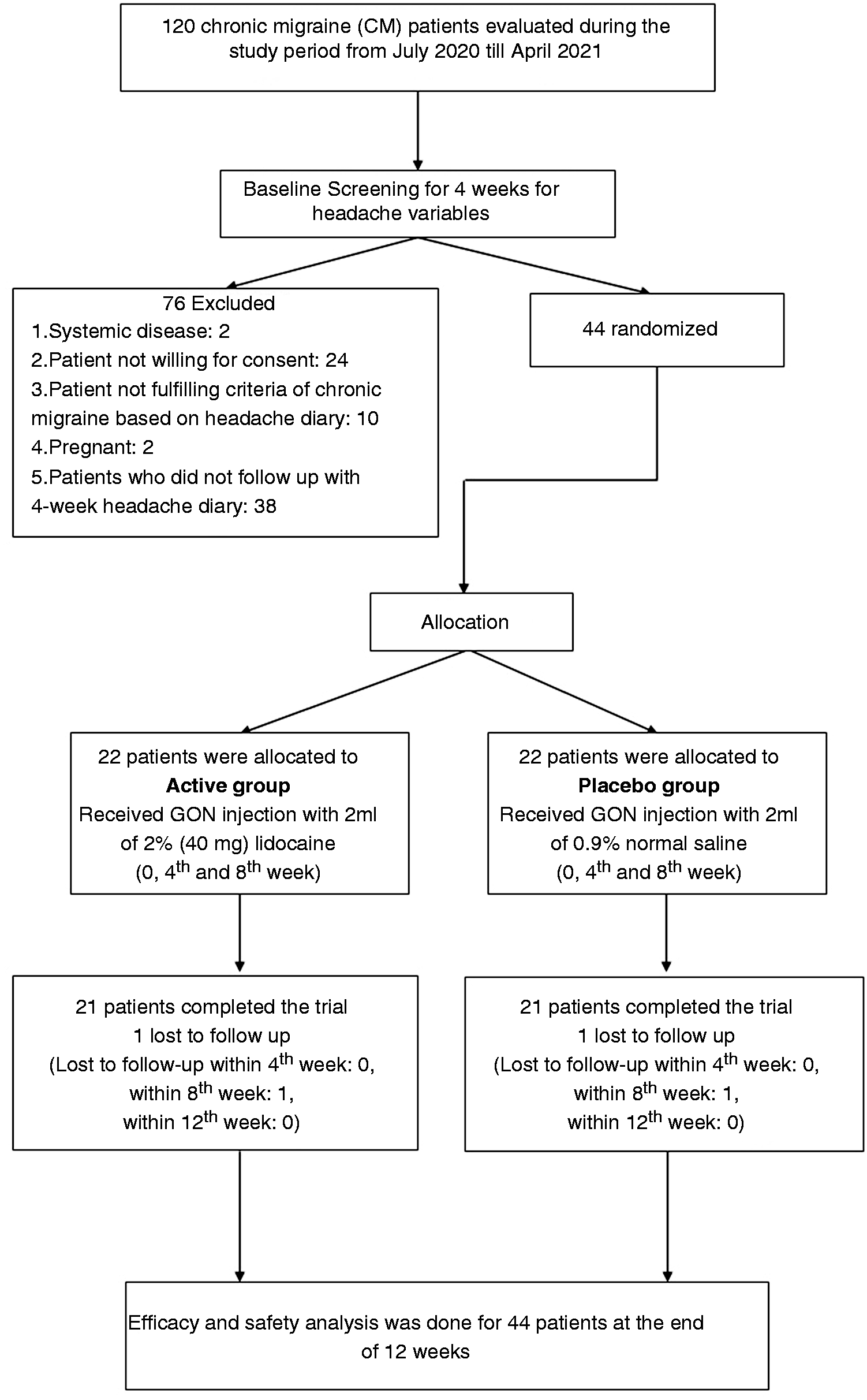
Flow of the study.
Baseline characteristics of the patients, according to Trial Group.*

*Plus–minus values are means ± SD. The intention-to-treat population included all the patients who underwent randomization. Patients in the active group received greater occipital nerve injections of 2 ml of 2% lidocaine and those in the placebo group received 2 ml of 0/9% saline at baseline and weeks 4 and 8. There were no significant between-group differences at baseline for any characteristic. **Psychiatric co-morbidities namely depression and anxiety were measured by Hamilton depression scale and Generalized anxiety disorder assessemnt-7 scale for respectively. §A headache day was defined as a calendar day in which headache pain lasted at least 4 consecutive hours and had a peak severity of at least a moderate level, or a day in which acute migraine–specific medication (triptans or ergots) was used to treat a headache of any severity or duration. ¶A migraine day was defined as a calendar day in which headache pain lasted at least 4 consecutive hours and met criteria for migraine (with or without aura) or probable migraine (subtype in which only one migraine criterion is absent), or a day in which acute migraine–specific medication (triptans or ergots) was used to treat a headache of any duration. €Acute migraine medication day included a day in which the patient consumed for the relief of headache of any duration and severity nonsteroidal anti-inflammatory agents, triptans, combination of naproxen and sumatriptan, or ergots. ¥Rated by the patients from 0–10 (a higher score denoting greater severity). £Calculated as total number of hours with any headache severity per day multiplied by headache days. ƛThe six-item Headache Impact Test (HIT-6) questionnaire assesses headache-related disability over the preceding 4 weeks, with scores ranging from 36 to 78 and with higher scores reflecting greater disability. ǂMigraine disability assessment scale (MIDAS), a five-item self-administered questionnaire with a higher score indicating greater disability. Baseline MIDAS was calculated based on 3 months data prior to randomization ϮA 3-dimension migraine specific quality-of-life questionnaire (MSQ) score with a higher score indicating better quality of life.
Efficacy
With respect to primary endpoint, change in mean number of headache days at 9–12 weeks compared with baseline was −7.2 days (95% CI: −10.9 to −5.8) for active group compared with −3 days (95%CI: −6.7 to −2.4) for placebo. The LSM difference between the two treatment groups was −4.2 days (95% CI: −7.5 to −0.8; p = 0.018) (Table 2, Figure 3A). Reduction in mean number of migraine days was greater for the active group (−6.4 days [95%CI: −9.8 to −5.8]) compared with placebo (−1.8 days [95%CI: −5.1 to −1.6]) with the LSM difference of −4.7 days (95%CI: −7.7 to −1.7; p = 0.003) (Table 2, Figure 3B). At 9–12 weeks compared with baseline, a significantly greater proportion of patients in the active group (40.9%) achieved ≥50% reduction in headache days as compared to placebo (9.1%) (p = 0.024) (Table 2, Figure 3C). All other secondary end points demonstrated significant superiority of the active group compared with placebo except for mean change in AMT days. The LSM difference between the active and the placebo groups for mean changes in AMT days was −3.0 days (95%CI: −7.2 to 1.1; p = 0.164), VAS score −0.9(95% CI: −1.7 to −0.2; p = 0.016), CHH −68.7 hours (95% CI: −113.9 to −23.3; p = 0.004), HIT-6 score −5.4 [95% CI: −8.9 to −1.9; p = 0.003), MIDAS score −13.0 (95% CI: −6.5 to −19.4; p < 0.001), MSQ score 6.8 (95% CI: 11.6 to 2.2; p = 0.005) and CGI-S score −0.6 (95% CI: −1.2 to −0.2; p = 0.008) (Table 2).
Primary and secondary endpoints in the modified intention-to-treat population.*

*Efficacy analyses were conducted in the modified intention-to-treat population, which included all randomly assigned patients who received at least one dose of a trial regimen and had at least 1 follow-up assessment. To control the type I statistical error rate at 0.05, a pre-planned hierarchical testing procedure was applied for primary and the two key secondary endpoints which are presented in the sequence in which they were evaluated. Baseline refers to the 28-day preintervention period unless otherwise indicated.

Primary and key secondary endpoints. (a) Change from baseline in the average number of headache days per 4-weeks during the 12-week period after the first dose of the trial regimen (primary end point). A headache day was defined as a calendar dayContinued.in which headache pain lasted at least 4 consecutive hours and had a peak severity of at least a moderate level, or a day in which acute migraine–specific medication (triptans or ergots) was used to treat a headache of any severity or duration. (b) Change from baseline in the average number of migraine days per 4-weeks during the 12-week period after the first dose of the trial regimen (1st key secondary end point). A migraine day was defined as a calendar day in which headache pain lasted at least 4 consecutive hours and met criteria for migraine (with or without aura) or probable migraine (subtype in which only one migraine criterion is absent), or a day in which acute migraine–specific medication (triptans or ergots) was used to treat a headache of any duration. I bars indicate standard errors and (c) Proportions of patients in each trial group achieving ≥50% reduction in headache days per 4-weeks from baseline during the 12-week period after the first dose of the trial regimen (2nd key secondary end point). Changes from baseline to weeks 4, 8, and 12 are part of exploratory analyses.
Safety
No SAE was noted during the study. Overall, 64 TEAE, mostly mild and transient were reported by 31 patients during the trial (16 patients in the active group and 15 patients in the placebo). The distribution of TEAE is shown in Table 3. No patient discontinued the trial due to poor tolerability. Similar rates of TEAE immediately following the GON injection were observed in both groups that included dizziness, vasovagal syncope, and transient minimal local site bleeding. Dizziness and vasovagal syncope occurred only during the first injections. Minimal local site bleeding was the most common TEAE that occurred in 14 patients in each group. Few self-limiting TEAE such as abdominal pain (1), dysphonia (1), and throat pain (1) that lasted for a few days were not considered to be related to the investigational drug.
Adverse events in the safety population, according to trial group.*

*Shown are data collected during the double-blind, placebo-controlled intervention period. The safety population included all the patients who underwent randomization and received at least one dose of a trial regimen. Only one patient each in both groups discontinued allocated trial regimen after first dose due to inability to visit the hospital during COVID-19 lockdown.
Discussion
This study showed that four-weekly GONB injections with lidocaine for 12 weeks in CM patients was superior to placebo in decreasing the average number of headache and migraine days (4.2 and 4.7 days respectively) at 9–12 weeks. All other secondary endpoints including achievement of ≥50% reduction in headache days from baseline demonstrated significant superiority for the active group compared with placebo except for mean change in AMT days.
This is the first study with a robust methodology and analysis that evaluated the long-term efficacy and tolerability of GONB in CM as per the norms laid down by IHS for such trials (24). Three previous placebo-controlled double-blind randomized trials (17–19) had methodological limitations such as patient blinding (25) and produced conflicting results (15). Importantly, in the present study, both active and placebo groups received local application of lidocaine jelly to produce numbness prior to GONB, thereby ensuring patient blinding. Of the three previously published placebo-controlled double-blind randomized trials, one study reported superior efficacy of GONB using bupivacaine compared with placebo in CM patients at one month (17) (following weekly injections of GONB for four weeks) with the continuation of efficacy for the next two months during the open-label phase (when both groups received bupivacaine); the other two studies could not find superior efficacy of GONB compared with placebo at one month (18,19); the first of these studies employed ultrasound-guided GONB only once (18) while the second study employed weekly injections of GONB for four weeks without any further injections (19); the latter study showed superior efficacy of GONB at the end of second and third month (19). Thus, there was significant heterogeneity amongst the previous studies in terms of design and methodology and the outcomes are difficult to interpret.
This study demonstrated that the effects of GONB by lidocaine accrue over time. A BOT-A trial for CM also showed that the effects of repeated injections accrue over time (26). The putative mechanism for such an effect may involve the modulation of the trigeminocervical pain pathway (12,13,27) rather than by the direct action of GONB in causing local anesthesia. In fact, a previous study showed that the effect of single GONB lasts far longer than its anesthetic effect (13). Though the exact mechanisms of such modulations remain unknown, a clinical study has shown a significant drop in interictal serum CGRP levels, a biomarker for migraine, after ultrasound-guided bilateral GONB compared to pre-blockade level (28). Similarly, improvement in neurophysiological correlates of CM following GONB has been reported (29).
The lack of statistically significant difference in efficacy measures between the active and placebo during the initial weeks of DBTP may be due to a large placebo effect, which has been noted in previous migraine preventive drug trials, more so for the invasive placebos such as injections (30). This argument partly explains the negative results of a trial of GONB by Dilli et al. (31) in a mixed cohort of episodic and CM patients in which 30% of patients of both active and placebo groups had ≥50% responder rate at the end of one month. Similarly, non-significant results between GONB and placebo at the end of one month were obtained in trials on patients with episodic migraine (32) and CM (18,19).
There was no significant difference in mean change in AMT days between the active group and placebo. It is possible that the patients were using acute migraine treatment even for low-intensity headaches; this was noted in some previous studies as well (33,34). Medication overuse at baseline was reported by 50% of patients receiving active treatment which was adjusted in the model for efficacy analysis. Thus, GONB seems to work well for CM patients with co-existent medication overuse. Favorable outcomes of GONB in chronic headache patients with medication overuse have been reported (35,36).
The group that received active treatment not only had greater improvement in patient-reported outcomes (PRO) such as HIT-6, MIDAS, and MSQ but also met the minimum cut-offs for clinically meaningful differences for these measures (37–39). Additionally, those receiving active treatment showed significantly greater improvement in CGI-S score which is a physician-determined summary measure of patient’s global functioning. Thus, the active treatment produced clinically meaningful effects.
GONB with lidocaine was well tolerated. Only minor and transient TEAE were reported. No SAE was reported. Except for injection-related TEAE, GONB with lidocaine did not produce any other significant systemic TEAE.
This study has several limitations. It recruited relatively younger female CM patients with a shorter duration of CM. Only four patients had a history of previous use of preventive medication, and none were taking preventive treatment at the time of randomization. Hence, this study population does not represent CM patients with a history of 2–4 preventive treatment failures. This may limit the generalizability of the study results. Finally, the study duration of only 12 weeks precluded any inference about the long-term effectiveness of the treatment. Despite these limitations, the findings of this study provide a new and cheaper alternative for the preventive treatment of CM that was found to be efficacious with good tolerability and safety profile.
Clinical implications
Four-weekly greater occipital nerve blockade with 2% lidocaine for 12 weeks was superior to placebo in decreasing the average number of headache and migraine days in patients with chronic migraine. Greater occipital nerve blockade with 2% lidocaine in patients with chronic migraine produced mild, transient, and self-limiting adverse effects. None of the patients discontinued the treatment due to adverse effects. Patient-reported outcomes such as improvements in functionality, disability, and quality of life were clinically meaningful.
Footnotes
Acknowledgements
We wish to thank our patients for their participation in the trial. We thank Debanjan Chowdhury a scholar in neuroscience, Heidelberg University, Germany for his kind help for statistical analysis.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.