
Abstract
Background
The Transient Receptor Potential Ankyrin 1 (TRPA1) channel might play a role in migraine. However, different mechanisms for this have been suggested. The purpose of our study was to investigate the localization and significance of TRPA1 channels in rat pial and dural arteries.
Methods
Immunofluorescence microscopy was used to localize TRPA1 channels in dural arteries, pial arteries, dura mater and trigeminal ganglion. The genuine closed cranial window model was used to examine the effect of Na2S, a donor of the TRPA1 channel opener H2S, on the diameter of pial and dural arteries. Further, we performed blocking experiments with TRPA1 antagonist HC-030031, calcitonin gene-related peptide (CGRP) receptor antagonist olcegepant and KCa3.1 channel blocker TRAM-34.
Results
TRPA1 channels were localized to the endothelium of both dural and pial arteries and in nerve fibers in dura mater. Further, we found TRPA1 expression in the membrane of trigeminal ganglia neuronal cells, some of them also staining for CGRP. Na2S caused dilation of both dural and pial arteries. In dural arteries, this was inhibited by HC-030031 and olcegepant. In pial arteries, the dilation was inhibited by TRAM-34, suggesting involvement of the KCa3.1 channel.
Conclusion
Na2S causes a TRPA1- and CGRP-dependent dilation of dural arteries and a KCa3.1 channel-dependent dilation of pial arteries in rats.
Keywords
Introduction
The transient receptor potential ankyrin 1 (TRPA1) channel is a non-selective cation channel, which is involved in pain mechanisms (1,2). Specifically, it is suggested to play a role in the pathophysiology of migraine (3–5). This is based on the ability of migraine-causing substances to activate the TRPA1 channel and of migraine-preventive substances to inhibit the channel (6–9).
Different modes of action for the TRPA1 channel in migraine have been suggested. The TRPA1 channel is localized to dural afferent neurons, and activation of TRPA1 in these neurons causes release of calcitonin gene-related peptide (CGRP) (4,10–14). However, the TRPA1 channel has also been localized to the endothelium of pial arteries and a recent study showed that TRPA1 activation here causes vasodilation via opening of intermediate conductance Ca2+-activated K+ (KCa3.1) channels (15). Thus, both mechanisms are possible explanations for the role of the TRPA1 channel in migraine.
Activation of the TRPA1 channel in vivo can occur via environmental irritants such as umbellulone, formaldehyde or acrolein and via endogenous substances such as products of oxidative stress (7,16–18). Nitroxyl (HNO), which is the product of nitric oxide (NO) and hydrogen sulfide (H2S), activates TRPA1 leading to release of CGRP from dural neurons, trigeminal ganglion and brainstem (19,20). Thus, substances that are donors of either NO or H2S will also cause activation of the TRPA1 channel via formation of HNO.
In this study, we aimed to evaluate the effect of activation of the TRPA1 channel in pial and dural arteries by infusion of the H2S donor Na2S. Additionally, we examined the localization of the TRPA1 channel in rat pial and dural arteries, dura mater and trigeminal ganglion.
We hypothesized that Na2S causes vasodilation of both dural and pial arteries. Further, we hypothesized that the main effect in dural arteries is via release of CGRP from perivascular neurons, but that the vasodilation of pial arteries involves a contribution from the KCa3.1 channel.
Methods
Animals
Experiments were performed on 42 male Sprague Dawley rats (8–13 weeks; Taconic, Denmark). Rats were kept in a temperature-controlled room with a 12-hour light/dark cycle and food and water ad libitum. Experiments were approved by the Danish Animal Experiments Inspectorate (2014-15-0201-00256).
Immunofluorescence microscopy
A total of nine male rats (9–13 weeks) were used for immunofluorescence microscopy (IFM). We performed IFM on cross sections (pial artery, dural artery and trigeminal ganglion) and on whole mount preparations (pial artery and dura mater), where a branch of artery or piece of dura mater was placed on slides without sectioning. For preparation of arteries, the rats were euthanized either with CO2 with subsequent cervical dislocation or with an intraperitoneal injection (0.9 ml/kg) of a mixture of pentobarbital (200 mg/ml) and lidocaine (20 mg/ml) followed by decapitation. The middle cerebral and middle meningeal arteries, including smaller branches, were excised and pia mater was carefully removed from the middle cerebral artery. The arteries were placed in 2% paraformaldehyde for 15 minutes and were then transferred to 30% sucrose in phosphate buffered saline (PBS; Sigma-Aldrich, Steinheim, Germany) overnight. For preparation of the trigeminal ganglion and dura mater, rats were anaesthetized with pentobarbital as described above before transcardial perfusion with 300 ml cold PBS followed by 450 ml 4% paraformaldehyde (10°C). The trigeminal ganglion and dura mater were removed and placed in 4% paraformaldehyde for 1 hour followed by 10% sucrose for 24 hours, 20% sucrose for 24 hours and 30% sucrose for 3 days.
For cross sections, arteries and trigeminal ganglion were embedded in Tissue-Tek (O.C.T.™, Sakura, Belgium) and frozen in n-hexan (Sigma-Aldrich, Steinheim, Germany) submerged in liquid nitrogen. Cross sections of 5 µm were made with a cryostat (Leica CM 1950). The sections were then allowed to dry at room temperature for one hour and were stored at −20°C. The slides were rehydrated with PBS 3 × 5 minutes. A blocking step was performed by washing the slides for 50 minutes in PBS with 5% donkey serum, before incubating the slides with the primary anti-TRPA1 antibody (ACC-037, Alomone labs, Jerusalem, Israel; 1:200 in PBS with 1% bovine serum albumin) for 1 hour. Afterwards, the slides were washed with high-salt PBS (41.1 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4) for 5 minutes to reduce background staining followed by 2 × 5 minutes washes in PBS. The slides were then incubated with the secondary antibody (Alexa Fluor® 594 donkey anti-rabbit, Invitrogen, CA, USA; 1:800 in PBS) for one hour and washed with high-salt PBS and PBS again. Finally, DAPI (4′,6-diamidino-2-phenylindole, Life Technologies, Nærum, Denmark; 1:10.000 in PBS) was added for 30 minutes. Cover slips were mounted with Fluorescent Mounting Medium antifade agent (S3023) (Dako, Glostrup, Denmark).
For whole mount preparations, the staining was performed by transferring the tissues from one solution to another. Incubation times were the same as for cross sections. The tissues were incubated with primary antibodies raised against TRPA1 (ACC-037) and CGRP (C7113, Sigma-Aldrich, Steinheim, Germany). ACC-037 was diluted 1:200 for pial arteries and 1:400 for dura mater and C7113 was diluted 1:1000 for pial arteries and 1:500 for dura mater. The tissues were incubated with two secondary antibodies (Alexa Fluor® 594 donkey anti-rabbit, 1:800 and Alexa Fluor® 488 donkey anti-mouse, Invitrogen, CA, USA, 1:250). The pial artery and dura mater were placed on slides and cover slips were mounted.
The specificity of the primary TRPA1 antibody (ACC-037) was checked on cross sections of the dural and pial arteries by pre-incubation with the peptide antigen (Alomone labs, Israel). The pre-incubation experiments were done with vessels from three different animals using a primary antibody concentration of 4.25 μg/mL and a blocking peptide concentration of 6 μg/mL. Specificity of the TRPA1 antibody in dura mater and trigeminal ganglion was checked by omission of the primary antibody. Furthermore, the specificity of ACC-037 towards TRPA1 was recently validated using cardiomyocytes from TRPA1 knock-out mice (21).
The slides were viewed with an Olympus BX50-FLA fluorescence microscope (Olympus, Hamburg, Germany) using Type F Immersion Liquid (Leica Microsystems CMS GmbH, Wetzlar, Germany) in combination with 40 × (NA 1.00 Oil Iris) or 60 × (NA 1.40 Oil) Olympus UPlanApo objectives. Images were captured with Retiga™ 6000 scientific monochromatic CCD camera using Q-Capture Pro7 Software (both Teledyne QImaging, Surrey, Canada) to control the camera. The exact same microscope and camera settings, and the same amount of post-capture image processing (using Adobe Photoshop CS6) were used for documenting positive stainings and negative control stainings.
Closed cranial window model
Surgical procedure
Experiments were performed on a total of 33 rats (8–13 weeks). Rats were anaesthetized with an intraperitoneal injection (0.325 ml/kg body weight) of pentobarbital (200 mg/ml) and lidocaine (20 mg/ml). Anesthesia was maintained with iv infusion of pentobarbital (50 mg/ml in saline, 0.15–0.23 ml/hour). Body temperature was maintained with a rectal thermometer and a heating pad. The trachea was cannulated and connected to a ventilator (SAR-830/P Ventilator, CWE Inc. AD Instruments, Oxford, UK).
Catheters were placed in the femoral artery and vein on both sides (BTPE-10, Polyethylene tubing, .011 × .024in/.28 × .60 mm and BTPU-040, Polyurethane tubing, .025 × .040in/.63x1.02 mm). The veins were used for infusion of pentobarbital and experimental drugs. The arteries were used for continuous monitoring of blood pressure and for blood samples. Arterial blood samples were analyzed prior to the experimental protocol and at least once more halfway through the protocol. The ventilator was adjusted so that arterial gas tensions and pH were kept within the following ranges: pH 7.35–7.45; PaCO2 35.2–42.7 mmHg; PaO2 81.7–127.5 mmHg. A free-floating catheter was placed in the right carotid artery and the rat was placed in a stereotaxic frame (22). A cranial window was prepared by exposing the right parietal bone, covering it with saline and thinning it to transparency with a dental drill. To prevent the window from drying, it was covered in mineral oil. Branches of the middle cerebral artery and the middle meningeal artery were viewed with a stereomicroscope (Leica Model MZ 16; Leica Microsystems, Brønshøj, Denmark) connected to a digital camera (Kappa CF8/5; Kappa Optronics GmbH, Gleichen, Germany). Branches of the middle meningeal and middle cerebral artery located close to the border of the temporal bone were chosen. The diameter of the arteries was monitored with a video dimension analyzer (V94; Living Systems Instrumentation Inc., Burlington, VT, USA) allowing one dural and one pial artery to be measured in each animal along with recording of mean arterial blood pressure (MABP) using Perisoft software (Version 2.5.5; Perimed AB, Järfälla, Sweden).
Experimental protocol
Drugs were infused into the carotid artery (ic) or the femoral vein (iv) at a rate of 125 µl/kg/min. All drugs were given in a volume of 125 µl/kg except TRAM-34, which was given in a volume of 250 µl/kg. At the beginning of each experiment, saline was infused as a negative control and CGRP (500 ng/kg, ic) was infused as a positive control.
A dose-response experiment was performed initially to determine the doses of Na2S for the remaining experiments. Na2S was administered in stepwise increasing doses (0.05, 0.1, 0.3, 0.6, 1.2 and 2.3 mg/kg, ic).
The remaining rats were divided into three groups, receiving Na2S (ic) in combination with one of the following test substances: The TRPA1 antagonist HC-030031 (3 mg/kg, iv), the CGRP receptor antagonist olcegepant (100 µg/kg, iv), or the KCa3.1 channel blocker TRAM-34 (1 mg/kg, iv) (Figure 1). A dose of 2.3 mg/kg Na2S was chosen for the groups receiving HC-030031 and olcegepant. For the group receiving TRAM-34, a dose of 1.2 mg/kg Na2S was chosen as we anticipated this dose to be more selective for the endothelial response than for CGRP release from perivascular sensory neurons. Two consecutive doses of Na2S were given before administration of a test substance to ensure reproducibility. There were no significant differences (Dura; p = 0.39, pia; p = 0.43, MABP; p = 0.76) between the two applications of Na2S. The test substance was administered followed by a third and final dose of Na2S.

Timeline of the closed cranial window model. Saline and antagonists were administered five minutes before drug induced effects. Grey arrows indicate varying time between administrations depending on the time it takes for the response to return to baseline. Experiments performed with TRPA1 antagonist (HC-030031), CGRP receptor antagonist (olcegepant) or KCa3.1 channel blocker (TRAM-34).
Drugs
Na2S (Sigma Aldrich, Søborg, Denmark) was dissolved in nitrogen bubbled saline to a concentration of 18.4 mg/ml. A fresh solution was made on each experimental day and diluted with nitrogen bubbled saline for the final concentration. HC-030031, olcegepant and TRAM-34 (all from Tocris Bioscience, UK) were dissolved in Dimethyl sulfoxide (DMSO) to concentrations of 24 mg/ml, 10 mg/ml and 4 mg/ml, respectively. CGRP (Tocris Bioscience, Bristol, UK) was dissolved in saline to a concentration of 0.5 mg/ml. Olcegepant and CGRP were diluted with saline to the final concentration on each experimental day.
Statistical analysis
Artery diameter was measured in arbitrary units and MABP in mmHg. Peak values occurring 1 to 2 minutes after administration of the test substance were measured. Changes in dural artery diameter, pial artery diameter and MABP were used as parameters for effect of test substances. Change in diameter of arteries and MABP were calculated as percentage change from the baseline (the average of the 60 seconds preceding administration of the test substance). All values are presented as mean ± standard error of the mean (SEM). Effects of test substances were analyzed with one-way ANOVA test followed by Bonferroni’s multiple comparisons test for group-wise comparisons. p-values below 0.05 were considered significant. All analyses were made with GraphPad Prism 8.
Results
Immunofluorescence microscopy
We found strong expression of TRPA1-immunoreactivity (IR) in the endothelium of frozen cross sections of both dural and pial arteries (Figure 2(a)/(c)). In pial arteries, we also found TRPA1-IR in adventitia (Figure 2(c)). The sections pre-incubated with primary antibody together with its blocking peptide did not show any visible TRPA1 staining (Figure 2(b)/(d)). In whole mount preparations of pial artery and dura mater, we found scattered TRPA1-IR and nerve fibers showing CGRP-IR (Figure 3). The finding of TRPA1 staining in fiber-like structures in the whole-mount preparation of pial arteries (Figure 3(b)) is consistent with the TRPA1-IR we observed in adventitia of pial arteries in frozen cross sections (Figure 2(c)). In trigeminal ganglion, an abundance of TRPA1-IR was localized to a membrane-like structure, some of which in the outer lining of neurons expressing CGRP-IR (Figure 4). Co-localization of TRPA1 and CGRP staining was only seen at occasional spots at the borderline between the intracellular space and membrane of the TG neurons.
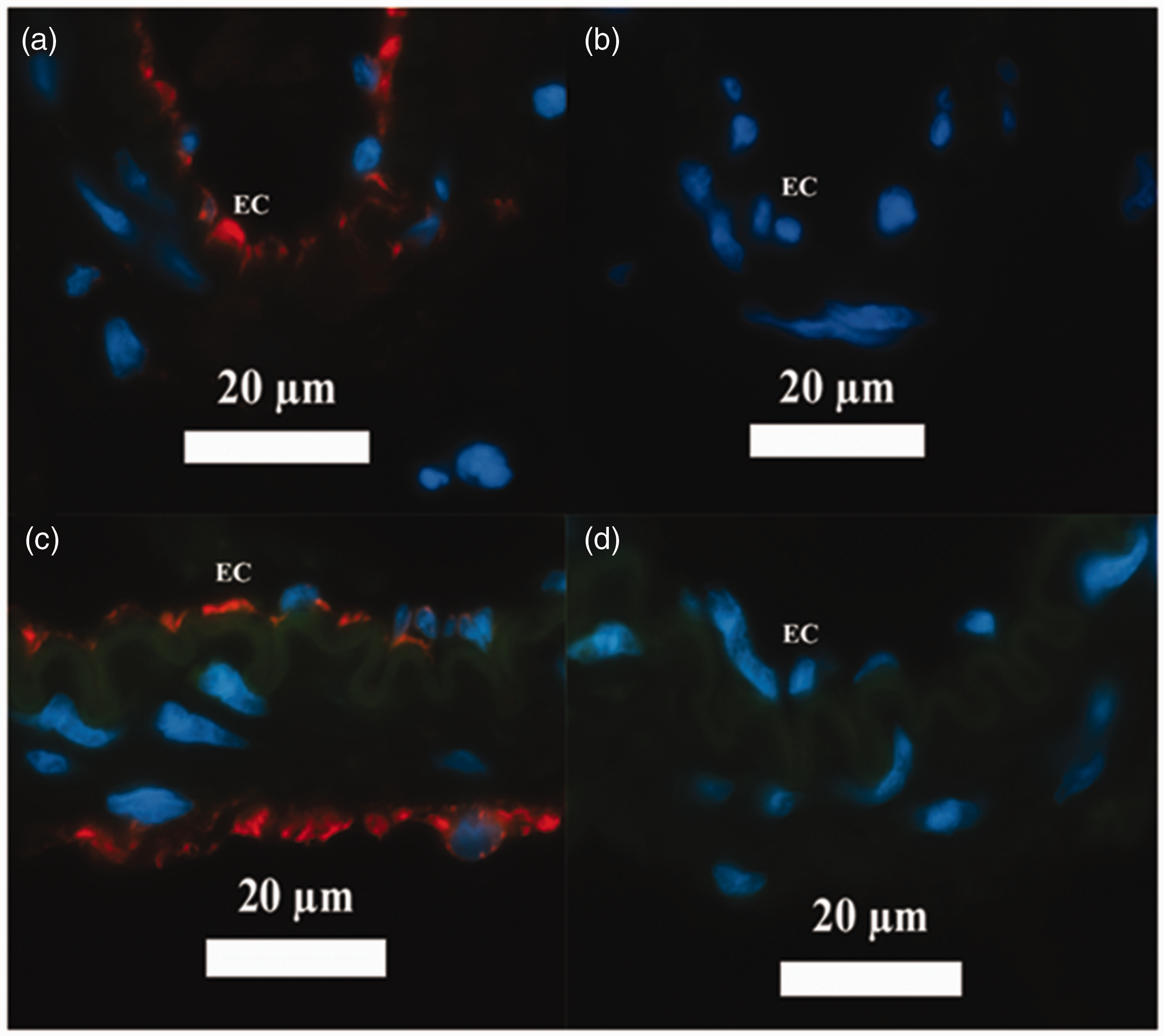
Immunofluorescent stainings of TRPA1 channels in rat dural and pial arteries. Red represents TRPA1, blue represents DAPI and green is the autofluorescence of the internal elastic lamina. (a) Dural artery with the primary TRPA1 antibody, (b) dural artery with TRPA1 antibody plus the blocking peptide, (c) pial artery with the primary TRPA1 antibody, (d) pial artery with TRPA1 antibody plus the blocking peptide.

Immunofluorescent stainings of TRPA1 channels and CGRP in whole mount preparations of (a) dura mater and (b) pial artery. Red represents TRPA1 channels, blue represents DAPI and green represents CGRP. In dura mater, green autofluorescence is also seen in a blood vessel.

Immunofluorescent staining of TRPA1 channels and CGRP in rat trigeminal ganglion. (a) With primary antibodies for TRPA1 channels, (b) CGRP and (c) overlay of TRPA1 and CGRP. The arrows show in yellow co-localization of CGRP and TRPA1 along the surface membrane of the neuronal cells. Red represents TRPA1 and green represents CGRP.
Closed cranial window model
Effect of CGRP and Na2S
Arterial dilations occurred within the first minute after administration of CGRP and Na2S. The duration of the dilation was 2–5 minutes for CGRP and less than 1 minute for Na2S. When administering increasing doses of Na2S, we found no significant effect of the low doses (0.05 mg/kg to 0.3 mg/kg) in the two vascular beds. In dural arteries, doses of 0.6 mg/kg, 1.2 mg/kg and 2.3 mg/kg caused significant dilations of 66.6 ± 12.6% (n = 5, p < 0.001), 85.3 ± 19.1% (n = 7, p < 0.0001) and 55.8 ± 10.8% (n = 8, p < 0.001), respectively (Figure 5, left). Correspondingly, the same three doses caused significant dilation of pial arteries of 49.7 ± 16.3% (n = 6, p < 0.005), 47.4 ± 8.2% (n = 8, p < 0.005) and 54.2 ± 16.8% (n = 7, p < 0.001; Figure 5, middle). Only at 2.3 mg/kg a significant drop in MABP of −22.6 ± 6.9% was observed as compared with vehicle (n = 10, p < 0.0001; Figure 5, right).
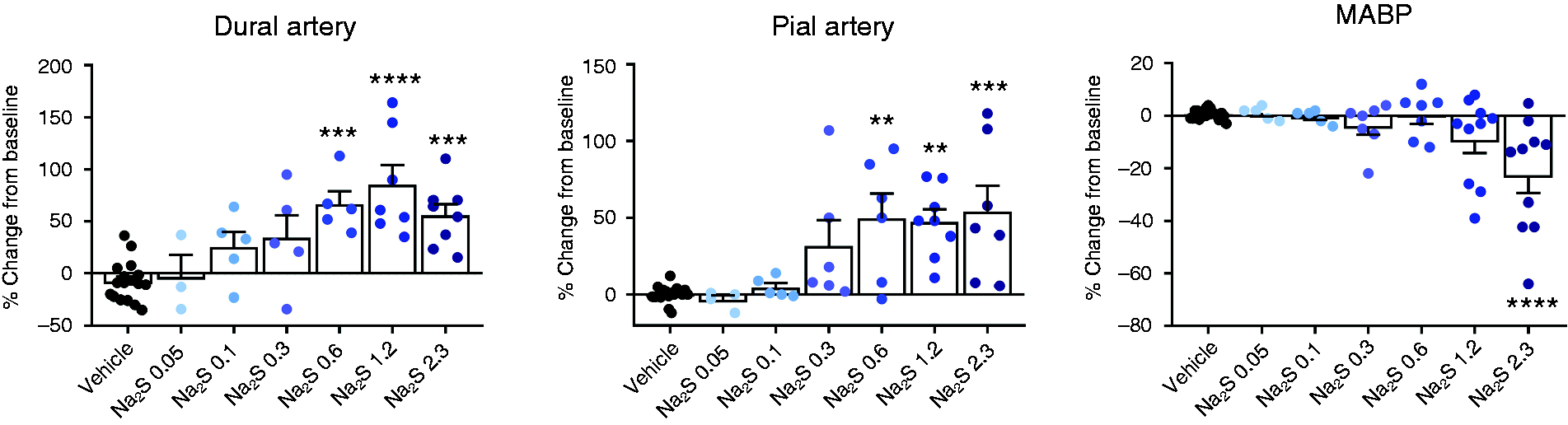
Effect of intracarotid administration of increasing doses in mg/kg of Na2S on the diameter of rat dural and pial arteries and MABP. The number of experiments is 3–20 as indicated by colored bullets in the figures.
When combining all subsequent experiments, 1.2 mg/kg and 2.3 mg/kg Na2S caused a dilation of dural arteries of 80.4 ± 20.5% (n = 6, p < 0.05) and 64.9 ± 10.6 (n = 20, p < 0.0001), respectively. CGRP caused a dilation of 77.0 ± 8.9% (n = 20, p < 0.0001; Figure 6). In pial arteries, 1.2 mg/kg and 2.3 mg/kg Na2S caused dilations of 51.3 ± 8.2% (n = 8, p < 0.01) and 47.4 ± 10.3% (n = 12, p < 0.01), respectively. The dilations induced by both concentrations of Na2S were significantly (p < 0.01) larger than the response to CGRP, which did not cause a significant change in diameter in pial arteries (−1.3 ± 2.9%, n = 20; Figure 6). Na2S doses of 1.2 mg/kg and 2.3 mg/kg Na2S and CGRP caused drops in MABP of −14.4 ± 7.1% (n = 8, p > 0.05), −16.1 ± 5.3% (n = 15, p < 0.01) and −21.7 ± 2.4% (n = 23, p < 0.0001), respectively (Figure 6).

Effect of intracarotid administration of Na2S and CGRP (500 ng/kg) on diameter of rat dural arteries, pial arteries, and mean arterial blood pressure. The number of experiments is 6–26 as indicated by colored bullets in the figures.
Effect of HC-030031, olcegepant and TRAM-34
Infusion of HC-030031, olcegepant or TRAM-34 did not cause any significant dilation or contraction of dural and pial arteries (p > 0.05).
Effect of HC-030031
In the experiments with in vivo administration of HC-030031, Na2S alone caused dilation of dural arteries of 55.8 ± 10.8% (p < 0.01 vs. vehicle). This was reduced to 14.6 ± 12.8% after administration of HC-030031 (n = 8, p < 0.05 vs. Na2S; Figure 7(a)). In pial arteries, the Na2S-induced dilation was 36.7 ± 14.7% (p > 0.05 vs. vehicle), and it was not significantly affected by administration of HC-030031 (21.7 ± 22.8%, n = 6; Figure 7(b)). MABP was changed non-significantly (−22.9 ± 8.4%) in response to Na2S infusion. After administration of HC-030031, the MABP response to Na2S was −5.7 ± 6.8% (n = 8).

Effect of HC-030031 (3 mg/kg) ((a) and (b)), olcegepant (100 μg/kg) ((c) and (d)) and TRAM-34 (1 mg/kg) ((e) and (f)) on Na2S-induced responses in rat dural arteries ((a),(c),(e)) and pial arteries ((b),(d),(f)). The number of experiments is 6–8 as indicated by colored bullets in the figures. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. corresponding vehicle; #p < 0.05 vs. previous administration of Na2S alone.
Effect of olcegepant
In the experiments with in vivo administration of olcegepant, Na2S alone caused dilation of dural arteries of 83.3 ± 17.6% (n = 7, p < 0.001). Administration of olcegepant caused a significant reduction of the dilation to 25.1 ± 10.6% (n = 7, p < 0.05; Figure 7(c)). In pial arteries, Na2S caused a dilation of 47.3 ± 13.5% (n = 6, p < 0.05) without olcegepant vs. 22.4 ± 11.7% with olcegepant (n = 6, p > 0.05; Figure 7(d)). The effect of olcegepant on Na2S-induced dilation was not significant (p > 0.05). The effect of Na2S on MABP was −8.3 ± 5.1% (n = 7, p > 0.05) before administration of olcegepant. After administration of olcegepant, Na2S caused a non-significant change in MABP of −3.4 ± 3.7% (n = 7).
Effect of TRAM-34
The Na2S response was 80.4 ± 20.5% in dural arteries (n = 6, p < 0.05; Figure 7(e)) and 56.0 ± 8.9% in pial arteries (n = 8, p < 0.01; Figure 7(f)). After administration of TRAM-34, the responses to Na2S were still significantly higher than vehicle in both vessel types (n = 6–8, p < 0.05). In pial, but not in dural arteries, TRAM-34 caused a significant inhibition of Na2S-induced dilation (p < 0.05; Figure 7(f)). The change in MABP was not significantly different from vehicle with TRAM-34 (n = 8).
Discussion
We localized TRPA1 channels to the endothelium of pial arteries, trigeminal ganglion and dura mater, which is in agreement with previous studies (10,12,23). Importantly, we also found TRPA1 channels to be localized to the endothelium of dural arteries, which potentially has an impact on the role of TRPA1 channels for regulation of meningeal blood flow. It is known that activation of TRPA1 channels in sensory nerve fibers innervating the dura mater cause release of CGRP (4,14,19). This led us to investigate possible co-localization of CGRP and TRPA1 channels. We found no evidence of co-localization of TRPA1 channels and CGRP in cerebral or meningeal arteries. However, trigeminal ganglia neurons were surrounded by TRPA1-IR, indicating its localization to the membrane of trigeminal ganglia neurons as previously described (24). Some of these neurons were labelled with CGRP-IR, suggesting localization of TRPA1 and CGRP within the same neuron.
In the genuine closed cranial window model of changes in blood vessel diameter, we use CGRP as an internal standard for the viability of the preparation. However, CGRP does not pass the blood-brain barrier and although CGRP is a potent vasodilator in vitro it does not reach the smooth muscle cells of the pial arteries in the window model. Our results are in accordance with previous studies in the same model, which found CGRP to dilate dural, but not pial arteries in doses not affecting the MABP. The ic route of infusion was developed to avoid dilation of pial arteries due to autoregulation activated by a drug-induced fall in MABP (25).
Intracarotid infusion of Na2S dilates both dural and pial arteries. This is in line with our previous study with the TRPA1 agonist allyl isothiocyanate (AITC) (26). Infusion of substances via the carotid artery allow a more accurate assessment of the effects of pharmacological substances on the cranial vasculature without inducing significant systemic effects (25). This was also demonstrated here, as a higher dose of Na2S was required to induce a significant change in MABP (Figure 5).
Vasodilation induced by Na2S was dose-dependent, but in pial arteries the maximum response was at a somewhat lower dose (0.6 mg/kg) as observed in dural arteries (1.2 mg/kg). Furthermore, at the highest Na2S dose (2.3 mg/kg), the response in dural arteries was lower than at 1.2 mg/kg Na2S. Although this difference is not significant it can be explained by the repeated dosing regimen as previously shown in vitro, where a significant decrease in CGRP release after several repeated administrations of capsaicin was found (22). This phenomenon was not seen in pial arteries, which further supports a larger portion of the response here to be mediated via another mechanism than via release of CGRP.
The response to Na2S in dural arteries was dependent on TRPA1 channels, since it was inhibited by the TRPA1 channel blocker HC-030031. The Na2S response in dural arteries was also significantly blocked by the CGRP receptor antagonist olcegepant. Thus, these data support that Na2S-induced dilation of dural arteries is mediated via CGRP release from dural perivascular nerve endings as previously reported for TRPA1 activating substances (Figure 8) (7,13,26). Interestingly, in vitro experiments showed that intraluminal perfusion with HC-030031 caused an increase in myogenic tone (27). However, in the present study, no change in dural or pial arterial diameter was observed when HC-030031 was infused in vivo. It is not known to what extent the TRPA1 channels localized in endothelium of dural arteries contribute to the Na2S-induced dilation. Most likely, they do not play a major role in this response, since we did not see any effects in dural arteries of infusion of TRAM-34, a blocker of intermediate-conductance Ca2+-activated K+ channels (KCa3.1), which are known to be involved in endothelium-dependent hyperpolarization (Figure 8) (27). However, also small-conductance calcium activated K+ channels (KCa2.3) in the endothelium are activated by TRPA1 channel stimulation (27). Thus, the contribution of endothelial TRPA1 channels in dural arteries cannot be excluded until resistance to blockade of both types of Ca2+-activated K+ channels has been demonstrated. In pial arteries, the effects of TRPA1 and CGRP inhibition were opposite to the dural arteries. In experiments with HC-030031, the Na2S-induced dilation of pial arteries was not significant when compared to vehicle, which might have caused too small a window in order to measure a possible sensitivity to the TRPA1 antagonist. In the next set of experiments, Na2S-induced pial artery dilation was significant and it was not blocked by olcegepant, as expected from previous studies (26). In the present study, we were limited by the poor solubility of HC-030031 and were therefore not able to explore effects of higher concentrations of the drug in pial arteries. However, the dose used was effective in dural arteries. In vitro studies in pial arteries showed TRPA1 activation to cause dilation via a mechanism mediated by KCa3.1 channels (23,27). A significant inhibition of Na2S-induced dilation of pial arteries by the KCa3.1 channel blocker TRAM-34 was observed, which suggests that the main effect of Na2S in pial arteries is via endothelial mechanisms.

TRPA1 channel-induced intracranial artery dilation has in separate studies been shown to be mediated via two different mechanisms (27,31). One is endothelium-dependent and the other acts via perivascular release of CGRP. In this study, Na2S was used to activate the TRPA1 channels subsequent to its reaction with H2O and NO to form HNO. HNO activates TRPA1 channels located to perivascular sensory nerve fibers. This leads to Ca2+-influx via TRPA1 and release of CGRP from the nerve fiber, causing activation of the GS/AC/cAMP pathway and subsequent vasodilatation (31). The present study provided support for this mode of action in dural arteries. In endothelial cells, TRPA1 channels are located together with KCa3.1 channels in myoendothelial projections (MEP) across the internal elastica laminae (IEL). According to one study, activation of TRPA1 causes increased concentrations of intracellular Ca2+, leading to activation of KCa3.1 channels, which hyperpolarizes the endothelial cell membrane potential. This hyperpolarization is conducted via MEPs to underlying vascular smooth muscle cells resulting in myocyte hyperpolarization and vasodilation. K+ ions accumulating upon KCa3.1 activation in the intercellular space can activate inward rectifier K+ channels on vascular smooth muscle cells to cause further hyperpolarization and vasodilation (27).
TRPA1 is possibly involved in the pathophysiology of migraine (3,5). However, the mechanism for this involvement needs further investigation. Our results support previous findings that this effect could be mediated by TRPA1 channels in dural afferents and subsequent release of CGRP.
The possible role of cerebral arteries in migraine must not be disregarded. Previously, it was shown that the diameter of the middle cerebral artery, but not of the middle meningeal artery or extracranial arteries, was increased on the pain side during a spontaneous migraine attack (28). Due to the blood-brain barrier, it has been difficult to study the role of the endogenous signalling molecules, used as migraine-provoking substances, on cerebral arteries in vivo. However, one of the most important tools for the induction of migraine is nitroglycerine, which via the release of NO causes a significant and more pronounced dilation of the middle cerebral artery in migraine patients than in episodic tension-type headache sufferers and healthy controls (29). This further indicate a possible contribution of migraine pain via cerebral arteries. The pathophysiological mechanisms of this effect have, however, not been fully dissected. Here we hypothesize that Na2S, after the reaction with endothelial NO in cerebral arteries to form HNO, activates endothelial TRPA1 channels that partly, via the KCa3.1 channel, cause hyperpolarisation of vascular smooth muscle cells and thus dilation of cerebral arteries, which subsequently might be one out of many players in migraine pathophysiology (Figure 8). Whether CGRP is involved in the dilation of pial arteries after Na2S infusion cannot be determined as the CGRP-receptor antagonist olcegepant does not cross the blood-brain barrier (30).
In conclusion, we found TRPA1 channels to be localized to the endothelium of pial and dural arteries and to adventitia of pial arteries. Further, TRPA1 channels were localized in membrane-like structures surrounding CGRP-IR neurons in trigeminal ganglion. Activation of these channels after intracarotid infusion of Na2S causes dilation of both pial and dural arteries. In dural arteries, this dilation is mediated by TRPA1 channels and CGRP. In pial arteries, the dilation involves the KCa3.1 channel.
Article highlights
TRPA1 channels are localized to the endothelium of pial and dural arteries. Na2S causes dilation of dural arteries via activation of TRPA1 and CGRP. Na2S causes dilation of pial arteries that is dependent on the activation of the KCa3.1 channel.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by Candys foundation and the Danish Council for Independent Research (DFF-5053-00008).