
Abstract
Background
Efficacy and safety of erenumab have been evaluated in a comprehensive clinical development program resulting in approval for migraine prevention in over 40 countries to date.
Methods
This integrated safety analysis included four double-blind randomized trials and their extensions (up to three-plus years). Safety endpoints included exposure-adjusted patient incidences of adverse events, serious adverse events, and anti-erenumab antibodies.
Results
In all, 2375 of the patients randomized across the four studies received at least one dose of erenumab (70 mg or 140 mg), with cumulative exposure of 2641.2 patient-years. Exposure-adjusted adverse event rates during the double-blind treatment phase were similar to placebo, with the exception of injection-site reactions (17.1 vs. 10.8 per 100 patient-years), constipation (7.0 vs. 3.8 per 100 patient-years), and muscle spasm (2.3 vs. 1.2 per 100 patient-years). During the long-term extensions, adverse events reported were similar to those observed during the double-blind treatment phase, and rates of injection site reactions, constipation, and muscle spasm were reported at lower rates than in the double-blind treatment phase. There were two deaths reported, both confounded by pre-existing conditions.
Conclusions
This pooled safety analysis revealed a favorable and stable adverse event profile over time for erenumab with more than three years of exposure.
Trial registration
ClinicalTrials.gov NCT01952574, NCT02483585, NCT02456740, NCT02066415, and NCT02174861.
Introduction
Guidelines for the treatment of migraine recommend that patients with migraine featuring severe, disabling, or frequent attacks, as well as those who cannot tolerate or are nonresponsive to acute treatment, should receive preventive therapy aimed at decreasing the number, severity, and duration of attacks, improving responsiveness to acute medications, and improving function and quality of life (1–3). However, adherence to previously available preventive therapies is poor, attributable to many factors including lack of efficacy, poor tolerability, and possible interactions with other medications (4–6).
Erenumab (in the US, erenumab-aooe) is a fully human anti-calcitonin gene-related peptide (CGRP) receptor monoclonal antibody approved in the US and EU for migraine prevention (7,8). Regulatory approval was based on review of a comprehensive clinical trial program, including assessment of the safety and efficacy of multiple doses of erenumab versus placebo over a 3- to 6-month treatment period for the preventive treatment of episodic migraine (EM) or chronic migraine (CM) (8). These trials demonstrated robust efficacy across a broad population of patients with EM and CM, including those who are traditionally difficult to treat. Erenumab treatment resulted in significant reductions in monthly migraine days, increased proportion of patients with at least a 50% response rate, and reductions in migraine-specific acute medication use compared with placebo (9,10).
In addition to efficacy, the safety of newly emerging migraine preventive agents is of paramount importance, because any such agent may be used long-term and therefore must be both safe and well-tolerated to maximize adherence (11,12). Each of the trials individually showed a favorable safety and tolerability profile of erenumab in comparison with placebo (9,10,13,14). To gain further understanding of the safety profile of erenumab, we assessed integrated safety data across the pooled EM and CM prevention trials, both during the short-term (12–24 weeks) placebo-controlled phases as well as the longer-term active-treatment and open-label extensions. Including data across the broad development program enables a highly powered overview of the overall safety and tolerability profiles of erenumab in patients with migraine.
Methods
Patients
Summary of studies included in the pooled analysis.
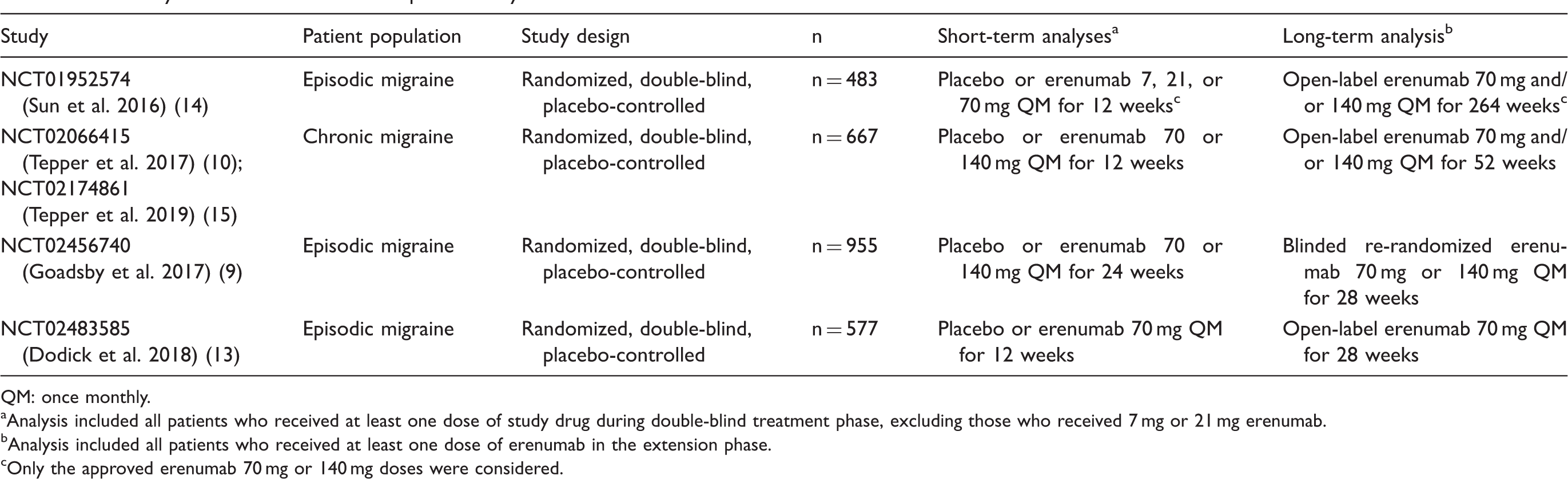
QM: once monthly.
Analysis included all patients who received at least one dose of study drug during double-blind treatment phase, excluding those who received 7 mg or 21 mg erenumab.
Analysis included all patients who received at least one dose of erenumab in the extension phase.
Only the approved erenumab 70 mg or 140 mg doses were considered.
Ethical considerations
Studies were conducted in compliance with the Declaration of Helsinki, International Council for Harmonisation Guidelines for Good Clinical Practice, and local country regulations. The study protocols were approved by the Institutional Review Board or Independent Ethics Committee at each center. Patients provided written informed consent before study initiation. Site investigators collected the data and Amgen conducted the data analyses. All authors interpreted the data and collaborated in manuscript preparation with support from a professional medical writer, funded by Amgen. All authors made the decision to submit the manuscript and attested to the veracity and completeness of data and analyses for their respective studies and the fidelity of this report.
Data sources
This was an integrated safety analysis conducted to assess the safety profile of erenumab with a larger number of exposed patients, providing more power and insight beyond that of individual studies assessed in isolation. This integrated analysis included four double-blind randomized trials (12 or 24 weeks in duration) and extension phases with varying erenumab dosage regimens and treatment durations (Table 1). All studies consisted of a screening visit, 4-week baseline phase, 12- or 24-week double-blind placebo-controlled treatment phase (DBTP), an extension phase comprising an open-label treatment phase (OLTP) or dose-blinded active treatment phase (ATP) ranging from 28 weeks to 5 years (Table 1). Safety follow-up visits were required 12–16 weeks after the last dose of erenumab, which may occur during the DBTP or extension phase. Two studies of EM (NCT01952574 and NCT02483585) had a 12-week DBTP and an OLTP (13,14); one study of EM (NCT02456740) had a 24-week DBTP and a 28-week re-randomized dose-blinded ATP (9); and one study of CM (NCT02066415) had a 12-week DBTP (10). Patients who completed the CM study DBTP could enroll in a 1-year OLTP under a separate protocol (NCT02174861) (15). All studies were completed at the time of analysis except for one (NCT01952574), for which an interim analysis was conducted at year 3 of the 5-year open-label extension. In each study, erenumab or matching placebo was administered by once-monthly (every 4 weeks) subcutaneous injection.
Safety endpoints and analysis
Safety endpoints included patient incidences of adverse events (AEs), serious adverse events (SAEs), and anti-erenumab antibodies (anti-drug antibodies; ADAs). Data were collected for all AEs and SAEs, coded using the Medical Dictionary for Regulatory Activities (MedDRA) version current at the time of analysis (V20.0), and graded according to Common Terminology Criteria for Adverse Events Version 4.03. Potential hypersensitivity reactions were identified using the hypersensitivity standardized MedDRA query (SMQ). Potential injection-site reactions (ISRs) were identified using preferred terms consistent with ISRs from the administration-site reactions and ISR high-level terms. Exposure-adjusted incidence rates (per 100 patient-years of follow-up) for AEs and SAEs were calculated by preferred term.
Immunogenicity of erenumab was evaluated using an electrochemiluminescence-based bridging immunoassay for the detection of ADAs. For patients whose sera tested positive in the immunoassay, an in vitro biological assay was performed to detect neutralizing antibodies.
Safety population and statistical analyses
The safety analysis sets included all patients from four studies who received at least one approved erenumab dose (70 mg or 140 mg) or placebo during the DBTP (short-term analysis) or at least one dose of erenumab (70 mg or 140 mg) during the extension periods (long-term analysis) per standard methods analyzing safety outcomes for patients receiving ≥1 dose of study therapy as treated (Safety Population) (16). Including all patients exposed during the clinical trials captures the full extent of the AE rates as experienced in these studies with both short- and long-term exposure.
For the long-term analysis, because the erenumab dose for some patients could have been increased from 70 mg to 140 mg during the extension periods, these patients were counted in both dose groups; however, the AE was ascribed to the dose the patients received at the time the event occurred. Exposure-adjusted analyses by dose received accommodated the sequential dose increase from 70 mg to 140 mg and differential patient exposure times, normalizing incidence rate to allow for rate comparison between short- and long-term analyses.
For the short-term analyses, events with onset dates on or after the first dose of study drug during DBTP were included. For the long-term analysis, events with onset dates after the first dose of erenumab during the extension (OLTP or ATP) were included. Events spanning the DBTP and extension phase were counted as a single occurrence in the short-term period unless there was a worsening of the event in the extension phase.
Exposure-adjusted incidence rates (per 100 patient-years) of AEs during DBTP or extension were calculated by taking the total number of patients who reported at least one event, dividing by total time at risk for reporting the AEs (in years, summed across all patients), and multiplying by 100. Time at risk only included time to first occurrence of the AE. If a patient did not have the corresponding AE to report, then the time at risk included time to first dose in extension phase for short-term analysis or safety follow-up visit for long-term analysis. Incidence of patients who developed anti-erenumab antibodies at any time was tabulated.
Results
Short-term analysis during double-blind treatment phase
Patient demographics, disposition, and exposure
Demographics.
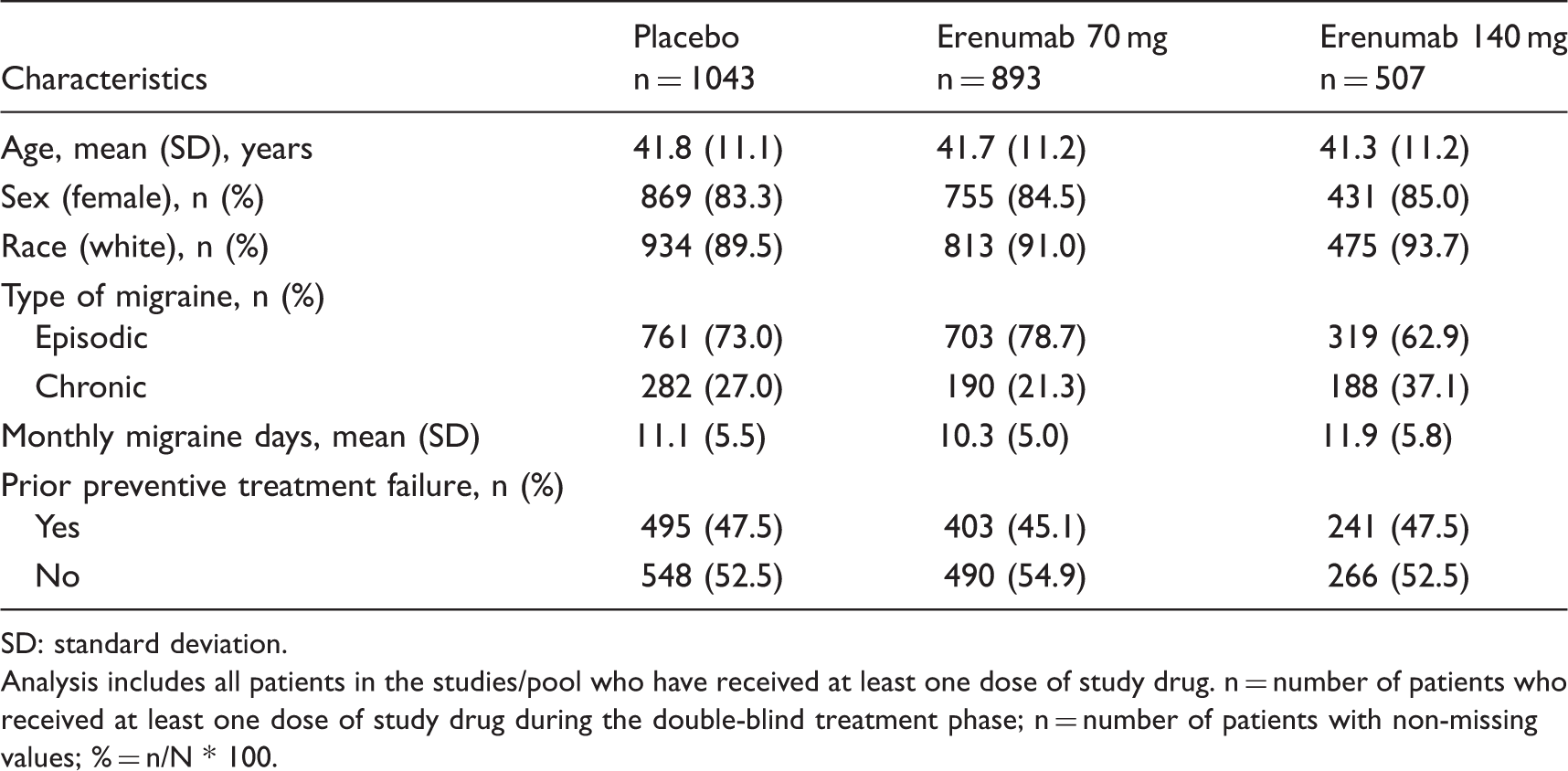
SD: standard deviation.
Analysis includes all patients in the studies/pool who have received at least one dose of study drug. n = number of patients who received at least one dose of study drug during the double-blind treatment phase; n = number of patients with non-missing values; % = n/N * 100.
Patient disposition.

Safety analysis
Exposure-adjusted incidence of adverse events.

AE: adverse event; CTCAE: Common Terminology Criteria for Adverse Events; MedDRA: Medical Dictionary for Regulatory Activities; pt-yr: patient-years; n: number of patients who received at least one dose of erenumab or placebo; n: number of patients reporting at least one occurrence of an adverse events; e: sum across all patients, the total time at risk in years; r: exposure-adjusted patient incidence rate per 100 pt-yr (n/e * 100).
Note: Most common adverse events had an incidence of ≥3.0 per 100 pt-yr in patients treated with erenumab during the DBTP or long-term extension phase. Grading categories were determined using CTCAE version 4.03.
Events coded as nasopharyngitis in prior MedDRA versions were recoded to viral upper respiratory tract infection in MedDRA v20.0.
There were no injection-site reactions of grade ≥3 reported.
Hypersensitivity reactions were identified using the hypersensitivity standardized MedDRA query.
The AEs that occurred with an incidence rate ≥4 patients per 100 patient-years greater than placebo for either dose of erenumab during the DBTP analysis included constipation, muscle spasms, and those related to ISRs. Exposure-adjusted rates of constipation were 10.7 per 100 patient-years in the erenumab 140 mg group, 4.6 per 100 patient-years in the erenumab 70 mg group, and 3.8 per 100 patient-years in the placebo group (Table 4). Constipation was generally mild to moderate in severity and no serious event was reported. During the DBTP, constipation typically occurred early after initiating treatment (Supplemental Table 2). There was no pattern of gastrointestinal or constipation history in patients who developed constipation in the trials.
Exposure-adjusted incidence rates of muscle spasms per 100 patient-years were 5.2 in the erenumab 140 mg group, 0.4 in the 70 mg group, and 1.2 in the placebo group. Where location information was available, there was no pattern for the muscle spasms/cramps; most were reported in the leg, neck, and back.
The exposure-adjusted incidence rates of overall ISRs per 100 patient-years were 13.5, 19.6, and 10.8 in the erenumab 140 mg, erenumab 70 mg, and placebo groups, respectively (Table 4). The most frequent ISRs were injection-site pain, injection-site erythema, and injection-site pruritus. In the erenumab 140 mg, erenumab 70 mg, and placebo groups, respectively, exposure-adjusted incidence rates of injection-site erythema were 5.8, 3.5, and 0.9 per 100 patient-years; exposure-adjusted incidence rates of injection-site pain were 4.7, 13.0, and 5.7 per 100 patient-years; and exposure-adjusted incidence rates of injection-site pruritis were 2.1, 1.8, and 0.9 per 100 patient-years. Most ISRs were mild (Grade 1) and no ISR of Grade 3 or greater was reported.
There were no imbalances in hypersensitivity reactions. AEs mapping to hypersensitivity SMQ were low and were reported at a rate of 7.8 per 100 patient-years in the erenumab 70 mg group, 6.8 per 100 patient-years in the erenumab 140 mg group, and 7.0 per 100 patient-years of patients in the placebo group, (Table 4). The most frequently reported (>2 patients) preferred terms mapping to the hypersensitivity SMQ were rash, rash maculo-papular, and eczema. All other events were reported in ≤2 patients. None of the AEs of hypersensitivity reported in erenumab-treated patients were serious; two SAEs of hypersensitivity were reported in the placebo group. Only one case of a nonserious event of an anaphylactic reaction (140 mg group) was identified by the anaphylaxis SMQ among erenumab-treated patients in the DBTP. This event had an alternative cause (penicillin allergy) and was resolved following medical treatment. Thus, the AE was not attributable to erenumab, and the patient continued erenumab treatment without dosage alteration.
Immunogenicity
The incidence of binding ADAs during the DBTPs was low, with only a small subset of patients having neutralizing antibodies (Supplemental Table 3). The incidence of binding ADA development during the DBTPs was 6.3% (56/885) in patients receiving 70 mg erenumab (three of whom had in vitro neutralizing activity) and 2.6% (13/504) in patients receiving 140 mg erenumab (none of whom had in vitro neutralizing activity). In addition, two patients (0.1%) had pre-existing binding antibodies (non-neutralizing) at baseline prior to the first erenumab dose.
Long-term analysis during extension phase
Patient disposition and exposure
The long-term analysis involved 2375 patients who received at least one dose of erenumab 70 mg or 140 mg during the extension phases (OLTP or ATP) and includes patients in the 7 mg or 21 mg groups from the DBTP who received 70 mg or 140 mg during the extension phases. The cumulative total duration of exposure for patients in the long-term analysis was 2191.3 patient-years. Discontinuation rates remained low throughout the long-term extensions (Table 3); 418 (17.6%) patients discontinued the study drug during the extension phases and 65 (2.7%) cited an AE as the reason for discontinuation.
Safety analysis
Overall exposure-adjusted AE rates during the extension phases were similar to or lower than those observed during the DBTP, with no increase in rates, dose dependence, or suggestion of new safety concerns with longer-term treatment (Table 4). The types and natures of AEs and the incidence rates were comparable with previous observations and did not reveal any new safety concerns. The most frequent AEs in the erenumab groups were events commonly observed in clinical studies (e.g. nasopharyngitis/upper respiratory tract infection, nausea, sinusitis, and influenza) and were reported at similar rates in the placebo group during the DBTP.
Exposure-adjusted rates of AEs related to constipation, muscle spasms, and ISRs remained low (Table 4). Overall, rates of constipation of 1.6 per 100 patient-years in the long-term analysis were lower than those reported in the DBTP (Table 4 and Supplemental Table 2). Constipation was generally mild to moderate in severity and easily managed; no serious event was reported. Exposure-adjusted incidence rates of muscle spasms per 100 patient-years in the long-term analysis were 0.7 in the erenumab 140 mg group and 0.5 in the erenumab 70 mg group. The exposure-adjusted incidence rates of overall ISRs per 100 patient-years in the long-term analysis were 5.2 in the erenumab 140 mg and 5.6 in the erenumab 70 mg groups (Table 4).
There were two deaths reported during the long-term extensions; both were confounded by pre-existing cardiovascular risk factors and have been previously reported (17,18).
AEs mapping to hypersensitivity SMQ were lower than those observed during the short-term analyses and were reported at rates of 5.1 per 100 patient-years of patients in the erenumab 70 mg group and 3.7 per 100 patient-years in the erenumab 140 mg group (Table 4). The most frequently reported (≥10 patients) preferred terms mapping to the hypersensitivity SMQ in the long-term analysis were rash, urticaria, eczema, and hypersensitivity. None of the events were reported as serious. In the long-term analysis, the anaphylaxis SMQ identified four nonserious events in the erenumab-treated groups during the extension phases. Two of the events had alternative causes of their respective reactions (stinging insect and food allergy), and both resolved following medical treatment, with no alteration to erenumab dosage. Thus, these events were not attributable to erenumab. One of the additional events was reported as acute urticaria and dyspnea that occurred 4 days following erenumab 140 mg administration. The event resolved the next day following medical treatment and the patient discontinued erenumab treatment. The other event was cough and pruritus associated with a sore throat, laryngitis, sinus congestion, and generalized body aches that were reported 17 days following erenumab 140 mg administration. The event resolved following medical treatment, with no alteration to erenumab dosage.
Immunogenicity
The incidence of ADAs in the long-term analysis was low and had a high reversion rate (Supplemental Table 4). The incidence of binding ADA development was 8.0% (184 of 2300) in patients treated with erenumab (eight of whom had in vitro neutralizing activity). Most ADA responses were transient, 51.1% (94/184) of patients who developed binding ADAs and 75.0% (6/8) of patients who developed neutralizing antibodies reverted to a negative status by the last on-study time point (≤3.5 years). Of the two patients who remained neutralizing antibody–positive, one refused to participate in antibody follow-up tests and one withdrew from the study. ADAs developed as early as 2–4 weeks after the first dose; the majority developed within the first 6 months and very few after the first year. No ADAs developed after 2 years based on the current long-term analysis. There was no evidence of an impact of ADAs on drug efficacy and safety (including hypersensitivity SMQ, immune-related disorders system order class, and ISR SMQ) (Supplementary Table 5).
Discussion
This integrated safety analysis of over 3 years of erenumab treatment represents the longest-term data for a CGRP pathway-targeted therapy to date and allows for a more comprehensive assessment of the risk-benefit profile of erenumab treatment. Pooling data from multiple studies enhances the ability to identify less common events that may not be detectable in single studies. The most common events and overall rates of AEs and SAEs were consistent across the individual studies, implying that the studies were not heterogenous with respect to AE profiles. Data from the integrated safety analyses suggest no new safety signals with erenumab therapy for more than 3 years in this patient population beyond the safety profile described in the existing product label (7). The data were consistent with individual study results, in which erenumab was demonstrated to be generally well-tolerated in a large population of patients with migraine (9,10,13,14). With longer-term exposure for more than 3 years, there was neither an increase in AEs over time nor an occurrence of new AEs. There were no other apparent dose-related toxicities, and overall incidence rates of AEs, including hypersensitivity- and ISR-related AEs, were similar among treatment groups and similar to placebo. Incidence rates overall and for SAEs and AEs leading to treatment discontinuation were not increased with longer-term treatment.
During the erenumab development program, it was considered that inhibition of the vasodilatory effect of CGRP might present a theoretical risk of cardiovascular events (19,20). However, there was no observed AE signal related to cardiovascular events. Although these clinical trials excluded patients with existing cardiovascular disease, the findings are consistent with dedicated preclinical and clinical studies that collectively showed no evidence for negative vascular effects in the setting of CGRP receptor antagonism with erenumab (21–25). A detailed evaluation of the cardiovascular safety profile of erenumab (related to data pooled from these studies) is the subject of a separate analysis (26). There is no evidence to date to suggest that inhibition of the CGRP receptor poses a cardiovascular risk. No amount of clinical trial data obtained before marketing approval will ever elucidate all the information about potential risks; therefore, additional studies and post-marketing surveillance are essential to further refine the safety profile of erenumab, including safety data from patients not included in clinical trials (e.g. patients with comorbid cardiovascular conditions).
Given that the gastrointestinal tract is highly innervated by CGRPergic fibers and that CGRP can mediate gastrointestinal motility in some, but not all, animal models, blocking CGRP function, at least in theory, might alter gastrointestinal motility (27–29). In this pooled analysis, there was dose-related imbalance in incidence rates of constipation during the DBTPs consistent with the rates of 1% and 3% reported in the erenumab prescribing information label (7,8). This dose-related imbalance was not observed in the long-term treatment, where incidence rates for both doses of erenumab were lower than that reported for placebo during the DBTPs. Overall, the events in the erenumab studies were of mild to moderate severity (no event was serious), easily managed, alleviated with conservative treatment, and transient with continued erenumab treatment. Where information was available for patients in clinical trials experiencing constipation events, treatments were conservative and included laxatives, stimulants of bowel motility, probiotics, or dietary modification. That only one patient discontinued treatment due to constipation (on day 2 of treatment) indicates that these treatments allowed patients to successfully manage their constipation and continue erenumab treatment. Given the small number of constipation events in the pooled clinical trials, the decreasing risk observed in the long-term treatment, and that gastrointestinal history was not collected in detail, it is difficult to make any definitive conclusions regarding the lack of a pattern of gastrointestinal or constipation history in patients who reported constipation AEs. Further, the interpretation of these findings is confounded by the increased incidence of constipation in individuals with migraine compared with the general population (30–32).
All therapeutic protein products have the potential to induce immune responses and the clinical consequences of such immune responses can range from no apparent effect to SAEs, and/or interference with patient safety and therapy efficacy. The incidence of developing anti-erenumab antibodies was low. This may be due in part to the fully human nature of erenumab, which is expected to be less immunogenic when compared to therapeutic antibodies containing nonhuman coding sequences. The majority of ADAs developed within the first 6 months of treatment and were transient in nature. Development of anti-erenumab antibodies did not increase with longer duration of erenumab administration. Erenumab treatment during these studies did not result in severe hypersensitivity reactions or anaphylaxis. There were no imbalances or increases over time in hypersensitivity reactions, and the reported anaphylaxis reactions had alternative causes of their respective reactions (penicillin allergy, stinging insect, and food allergy) and thus, were not attributable to erenumab. Although the available data are too limited to make definitive conclusions, partly due to the low immunogenicity rate, the efficacy and safety of erenumab were not affected by the presence of anti-erenumab antibodies based on integrated long-term immunogenicity impact analysis.
Exposure-adjusted AE incidence rates correct for differences in time on trial by normalizing the rates to per 100 patient-years (33). Time on trial in this analysis was mostly due to differences in study length by design of the individual studies and not differential drop-outs, such that patients who were exposed to erenumab for longer periods of time relative to others contributed more to the calculation of incidence rates. Any events occurring after 3 years would have been captured by observations in those patients that remained in the trial for more than 3 years. Restricting the exposure-adjusted analyses to those patients who used erenumab for a specific time would drastically reduce the sample size and underestimate AE rates by not accounting for AEs that preferentially tend to occur early after initiation of treatment. By study design, patients in the phase 2 EM study would have the longest follow-up compared to other studies which were of shorter duration. Although any event after year 1.25 would come from a single study, this does not imply missing data from the shorter studies. Further, per protocol patients were to return 12 or 16 weeks after the last dose of study drug for a follow up safety visit, so they would still be assessed for adverse events following erenumab discontinuation.
Although pooled analyses provide more power to detect rare events that may not be observed in single studies, there are still limitations to these analyses. Exposure-adjusted incidence rates assume a constant hazard rate across time and AE rates could be underestimated if they occur more frequently later on. This can be partially adjusted for by summarizing the events by time intervals to show that rates do not change over time, as we have shown for constipation events (Supplementary Table 2). Such analyses are also limited in the ability to compare with AE rates of other treatments, because differences in study design and enrolled patient populations make direct comparisons among clinical trials unreliable and potentially misleading. Differences in event reporting and data collection between clinical trials and clinical practice, which often lack routine or standardized adverse event monitoring, limit the ability to generalize the results to the real world.
Conclusions
This pooled safety analysis describes the erenumab safety profile across the migraine clinical program to September 2017, with >2300 patients treated for up to 3 + years, representing the most comprehensive view of long-term safety with a CGRP-pathway inhibitor to date and reveals a favorable and stable AE profile for antagonism of the CGRP receptor with erenumab. Cardiovascular safety is being addressed separately (26). Overall, these analyses showed that there is no cardiovascular risk associated with erenumab use. In conclusion, erenumab demonstrated a favorable safety and tolerability profile, both in the short term as well as in the long term, supporting its use as a treatment for migraine prevention. Ongoing comparative clinical studies and postmarketing surveillance will provide further information on the erenumab benefit risk profile in real-world settings.
Supplemental Material
CEP888222 Supplemental Tables - Supplemental material for Long-term tolerability and nonvascular safety of erenumab, a novel calcitonin gene-related peptide receptor antagonist for prevention of migraine: A pooled analysis of four placebo-controlled trials with long-term extensions
Supplemental material, CEP888222 Supplemental Tables for Long-term tolerability and nonvascular safety of erenumab, a novel calcitonin gene-related peptide receptor antagonist for prevention of migraine: A pooled analysis of four placebo-controlled trials with long-term extensions by Messoud Ashina, David Kudrow, Uwe Reuter, David Dolezil, Stephen Silberstein, Stewart J Tepper, Fei Xue, Hernan Picard, Feng Zhang, Andrea Wang, Yanchen Zhou, Frank Hong, Jan Klatt and Daniel D Mikol in Cephalalgia
Footnotes
Clinical implications
Erenumab (in the US, erenumab-aooe) is a fully human anti-CGRP receptor monoclonal antibody approved in the US and EU for migraine prevention.
In pooled analysis of safety data over 3 + years, erenumab was found to be safe and well-tolerated with a spectrum and rate of adverse events consistent with the individual shorter-term placebo-controlled studies.
The favorable tolerability and safety profile, and the low discontinuation rates, suggest that adherence is favorable with erenumab and may result in positive long-term outcomes.
Acknowledgements
Erenumab is codeveloped in partnership with Amgen Inc. and Novartis. Jon Nilsen, PhD (Amgen Inc.), provided medical writing support for this manuscript.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MA: consultant or scientific advisor for Allergan, Amgen, Alder, Novartis, Teva, and Eli Lilly; primary investigator for Alder, Amgen, GM-11 gamma-Core-R, Novartis, and Teva trials; and grants from Lundbeck Foundation and Novo Nordisk Foundation. DK: consulting fees from Alder, Amgen, Biohaven, Eli Lilly, and Teva; and research investigator for Alder, Amgen, Biohaven, Eli Lilly, and Teva. UR: consulting fees, speaking/teaching fees, or research grants from Allergan, Amgen, Autonomic Technologies, CoLucid, ElectroCore, Eli Lilly, Medscape, Novartis, StreamMedUp, and Teva Pharmaceuticals. DD: consulting fees and speaking and/or teaching fees from Allergan, Amgen, Biogen Idec, Novartis, Bayer, and Teva. SS: consultant and/or advisory panel member for and/or honoraria from Alder, Allergan, Amgen, Avanir, Dr. Reddy’s, eNeura, ElectroCore Medical, Medscape, Medtronic, Mitsubishi Tanabe Pharma America, NINDS, Supernus, Trigemina, and Teva. SJT: employee of the Cleveland Clinic during this study; research grants (no personal compensation) from Alder, Allergan, Amgen, ATI, Dr. Reddy’s, ElectroCore, eNeura, Neurolief, Scion Neurostim, Teva, and Zosano; consultant and/or advisory boards fees from Acorda, Alder, Alexza, Allergan, AlphaSights, Amgen, ATI, Axsome Therapeutics, BioDelivery Sciences International, Biohaven, Cefaly, Charleston Labs, Decision Resources, DeepBench, Dr. Reddy’s, ElectroCore, Eli Lilly, eNeura, ExpertConnect, GLG, GSK, Guidepoint Global, Impel, M3 Global Research, Magellan Rx Management, Marcia Berenson Connected Research and Consulting, Medicxi, Navigant Consulting, Neurolief, Nordic BioTech, Novartis, Pfizer, Reckner Healthcare, Relevale, Satsuma, Scion NeuroStim, Slingshot Insights, Sorrento, Spherix Global Insights, Sudler and Hennessey, Supernus, Teva, Theranica, Thought Leader Select, Trinity Partners, Xoc, and Zosano; royalties from Springer; and salary from Dartmouth-Hitchcock Medical Center and American Headache Society. FX, HP, FZ, AW, YZ and DDM: employees of and stockholders in Amgen. FH and JK: employees of and stockholders in Novartis.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Amgen Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.