
Abstract
Introduction
Familial hemiplegic migraine 3 is an autosomal dominant headache disorder associated with aura and transient hemiparesis, caused by mutations of the neuronal voltage-gated sodium channel Nav1.1. While a gain-of function phenotype is generally assumed to underlie familial hemiplegic migraine, this has not been fully explored. Indeed, a major obstacle in studying in vitro neuronal sodium channels is the difficulty in propagating and mutagenizing expression plasmids containing their cDNAs.
The aim of this work was to study the functional effect of two previously uncharacterized hemiplegic migraine causing mutations, Leu1670Trp (L1670W) and Phe1774Ser (F1774S).
Methods
A novel SCN1A containing-plasmid was designed in silico and synthesized, and migraine mutations were inserted in this background. Whole-cell patch clamp was performed to investigate the functional properties of mutant Nav1.1 transiently expressed in Human Embryonic Kidney 293 cells.
Results and conclusions
We generated an optimized Nav1.1 expression plasmid that was extremely simple to handle and used the novel plasmid to study the functional effects of two migraine mutations. We observed that L1670W, but not F1774S, reduced current density and that both mutations led to a dramatic increase in persistent sodium currents, a depolarizing shift of the steady state-inactivation voltage-dependence, and a faster recovery from inactivation. The results are consistent with a major gain-of function effect underlying familial hemiplegic migraine 3. Our optimization strategy will help to characterize in an efficient manner the effect in vitro of mutations of neuronal voltage-gated sodium channels.
Introduction
Familial hemiplegic migraine (FHM) is a rare autosomal dominant disorder that is often associated with deeply debilitating symptoms like aura and transient hemiparesis. Mechanisms causing migraine in FHM might provide insights into more general processes that lead to common migraine. Three genes have been identified as causative agents for FHM1, FHM2, and FHM3, respectively, all of which are involved in membrane ion transport. FMH1 is caused by gain of function mutations in the neuronal presynaptic voltage gated Cav2.1 channel (1), whereas FHM2 is caused by loss of function mutations in the gene encoding the α2 subunit of the glia specific Na/K ATPase (2). More recently, SCN1A has been identified as the gene that is defective in FHM3 (3–5). SCN1A encodes the voltage-gated Nav1.1 channel, which is involved in generation and propagation of the action potential in CNS neurons and which is thought to be of particular importance in inhibitory interneurons (6). SCN1A is primarily known as a major target of epileptogenic mutations, most of which consist of missense and truncating mutations that lead to a loss of function of the channel (6–10). The epileptogenic phenotype observed in patients has been associated with a reduced excitability of GABAergic interneurons resulting in decreased inhibition of the neuronal network (6,11).
On the contrary, a gain of function mechanism is generally hypothesized in FHM3 based on the phenotype of FHM3 mutations in heterologous expression systems (3–5,12,13). The putative intensification of the inhibitory action of GABAergic neurons may lead to a general network silencing, which in turn could be the trigger event inducing cortical spreading depression (CSD), suspected to be of fundamental relevance in migraine with aura (14–17).
Until now, 12 SCN1A missense mutations have been described. Some of them (Q1489K, L1649Q, I1498M, F1661L, L1670W, and L1624P) (3,13,18) were identified in pure FHM families, whereas others were linked to FHM associated with either epileptic seizures (L263Q and T1174S) (19) or elicited repetitive daily blindness (Q1489H and F1499L) (20,21). In addition, F1774S, a de novo SCN1A mutation, was identified in a 19-year-old woman who had experienced episodes of hemiplegic migraine with aura since childhood. Sporadic hemiplegic migraine was diagnosed in the absence of family history (22).
Functional studies of Nav1.1 FHM3 mutations in heterologous systems have generated controversial results. For example, the L1649Q mutation was initially shown to slow the inactivation process when studied in the context of the cardiac SCN5A channel (5), but did not yield functional expression in HEK cells when studied in the neuronal SCN1A channel (23). More recently, Cestèle and collaborators succeeded in partially rescuing L1649Q expression by lowering cell incubation temperature or co-transfecting calmodulin and confirmed the slower kinetics of inactivation compared to WT (12). Electrophysiological studies on L1624P (13) and L263V (23) showed an overall gain of function as well. Conversely, investigations of mutations Q1489K and T1174S showed both gain and loss of function effects (23,24).
A major bottleneck in working with brain Nav channel plasmids has been the fact that these plasmids are technically challenging because they frequently undergo rearrangements during propagation and result in very low yield after purification. For the same reason, the efficiency of mutagenesis is usually extremely low (25).
Here, we devised a strategy to optimize the cDNA sequence of SCN1A using the highly homologous human cardiac SCN5A sequence as a template, and took advantage of the stability of the optimized plasmid to characterize by patch clamp technique two recently discovered migraine mutations, L1670W and F1774S (22,26) (Figure 1). Our results show that both mutations induce a dramatic increase of the “persistent” or “late” inward Na+ current, consistent with the hypothesis of a gain of function mechanism causing FHM3.
Structure of Nav1.1 and migraine mutations. The two investigated mutations (indicated by arrows) are mapped onto the cockroach Na channel structure (35). L1670W is located at the beginning of S5 of domain IV, while F1774S is located in the middle of S6 of domain IV. (a) lateral view with membrane indicated by gray lines; (b) different view in which only domain IV is shown fully. Voltage sensor segments, pore helices, S5 /S6 segments are colored differently. The figure was made with pymol.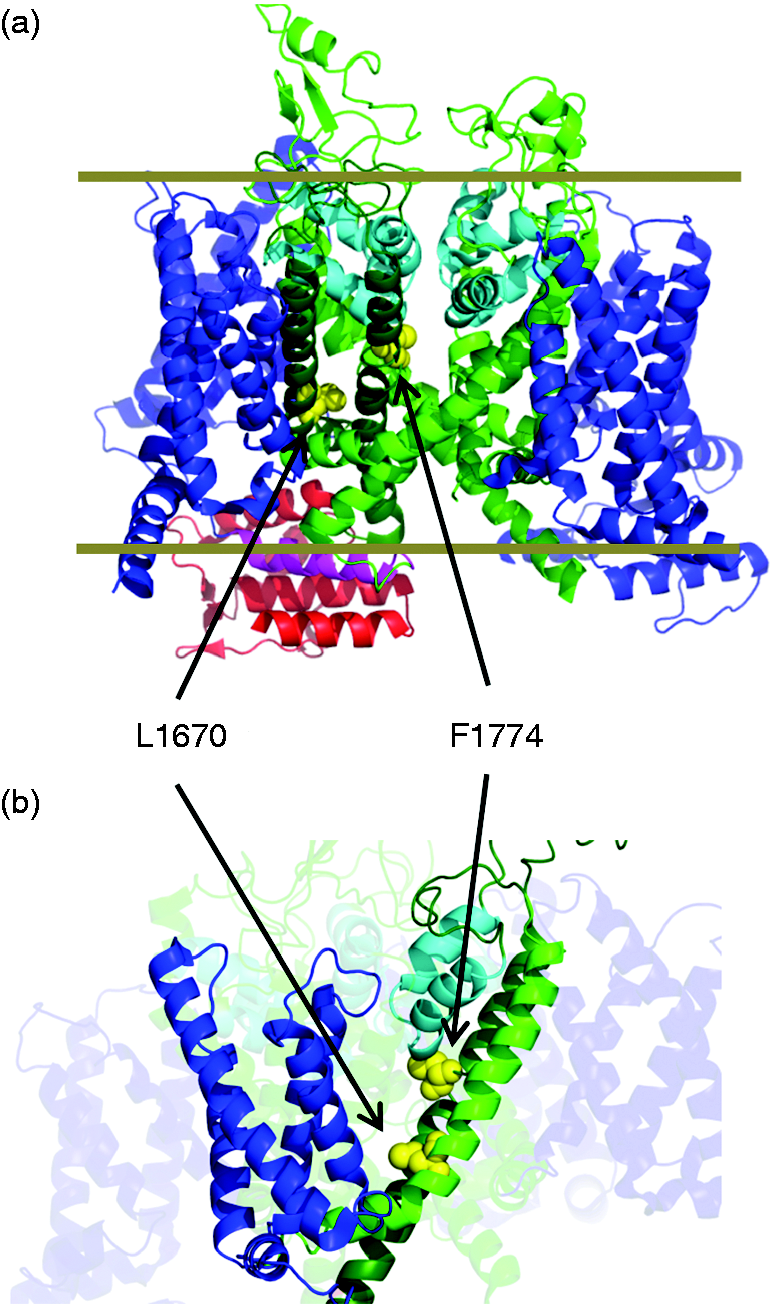
Materials and methods
Plasmid and mutagenesis
For the optimization, we used the shorter splice variant of the SCN1A cDNA, which encodes a 1998 amino-acid protein and which is supposed to be the predominant Nav1.1 variant expressed in the brain (27). This cDNA is shorter by 11 amino acids compared to the standard reference sequence (GenBank accession no. NM_006920.4). The in silico optimization procedure is described in Results. After the design was completed, the optimized cDNA was synthesized and sub-cloned into the pcDNA3.1 vector by GenScript (Piscataway, USA).
The original SCN1A plasmid was kindly provided by Dr. M Mantegazza and is described in reference (27). The mutations L1670W and F1774S were introduced into the optimized WT plasmid using restriction-free (RF) mutagenesis. The protocol consisted of 18 PCR cycles using a high fidelity Phusion DNA polymerase and 33 base long primers containing the desired mutation (see below). After digestion with DpnI, cDNA was transformed in standard DH5alpha bacteria, which were grown overnight at 37℃ on Ampicillin-containing agar plates. One clone for each mutation was amplified and fully sequenced. No spurious mutations were found.
The following primers were used for mutagenesis: L1670W: 5′ATGTCCCTGCCTGCCTGGTTCAACATCGGGCTG 3′ (forward) 5′ CAGCCCGATGTTGAACCAGGCAGGCAGGGACAT 3′ (reverse) F1774S: 5′ACATCATCATCTCCTCCCTCGTCGTGGTCAA 3′(forward) 5′TTGACCACGACGAGGGAGGAGATGATGATGT 3′ (reverse)
Cell culture
HEK 293 cells were grown in DMEM supplemented with 10% FBS, penicillin, and streptomycin and maintained at 37℃ in a 5% CO2, 100% humidity atmosphere. Cells were transfected by means of the Effectene Kit from Qiagen, using 600 ng of optimized SCN1A plasmid and 100 ng of a vector expressing GFP. Currents were recorded 24–48 h after transfection incubating cells at 30 or 37℃. Experiments were carried out in the absence of the β1 subunit because the functional effects of the β1 subunit are generally rather small (12). In addition, co-transfection of α and β subunits invariably leads to an uncontrolled variable stoichiometry, which might increase the variability of the data. Most importantly, the two mutations studied here are far away from the α-β interaction surface, as inferred from the eel structure (28). Thus, it is unlikely that they directly affect α-β interaction.
Electrophysiology
Sodium currents were recorded using the patch clamp technique in the whole-cell configuration as previously reported (5) using an Axopatch 200B amplifier and the GePulse acquisition program (http://users.ge.ibf.cnr.it/pusch/programs-mik.htm). Transfected cells were visualized by their fluorescence, and recordings were performed at room temperature (22–25℃).
Bath solution contained: 145 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Hepes), pH 7.4. Internal solution contained 40 mM CsCl, 10 mM NaCl, 80 mM CsF, 11 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 10 mM Hepes, 1 mM CaCl2, pH 7.3. Series resistance, RS, was between 2 and 4 MΩ, and the cell capacitance was between 10 and 25 pF, as measured by the compensating circuit of the amplifier. Maximum accepted voltage-clamp error calculated as Imax*RS was 5 mV. Data were analyzed with the program Ana (available at http://users.ge.ibf.cnr.it/pusch/programs-mik.htm) and Sigma Plot Systat Software, Inc. (SPSS) 2107, San Jose, CA, USA.
Various voltage protocols were applied to measure steady state and kinetic voltage dependent parameters; holding potential was −90 mV throughout. Leak and capacitive currents were subtracted using a P/4 protocol (except for the high frequency train stimulation).
The standard IV-protocol was performed by applying 20 ms long pulses to voltages ranging from −50 mV to +40 mV. The steady-state activation was determined by fitting the peak current-voltage (I-V) relationship with the equation:

A quantitative analysis of the time course of inactivation was performed by fitting the decaying phase of the sodium current with a double exponential function of the form:

These values were further combined to determine the relative area of the slow component of inactivation:

Steady-state inactivation was measured applying 100-ms conditioning pulses to various voltages followed by a test pulse to −10 mV, and currents were fitted with the equation:
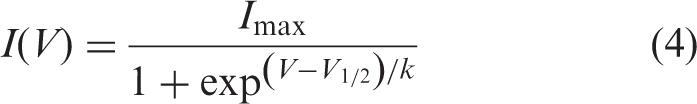
The time course of recovery from inactivation at −120 mV was measured by repolarizing the cell to −120 mV for a variable time after a 100 ms pulse to 0 mV, and assessing channel availability by a final test-pulse to −10 mV. Peak-currents at the final test pulse were fitted to a double exponential function:

Noise analysis: 200 pulses to 0 mV were applied. Non-stationary noise analysis (29) was performed essentially as described in (30). Baseline variance was subtracted, and the variance-mean plot was fitted by the equation:

Data are presented as mean ± standard error of the mean (SEM). Statistical significance was assessed using Student’s t test, and threshold p value for statistical significance levels set to *p < 0.05, **p < 0.01 and ***p < 0.001.
Results
Plasmid optimization strategy
Plasmids containing the cDNA sequence of the voltage gated sodium channel Nav1.1 (SCN1A) are notoriously difficult to amplify in bacteria and to mutagenize using PCR based or restriction-free mutagenesis, mainly because of a high rate of spontaneous recombination and low yield (25). The difficulties are only partially resolved by adopting specific conditions, such as a lower incubation temperature or special bacterial strains. Indeed, in a first attempt to introduce an FHM3 mutation in the original SCN1A plasmid using RF free cloning (see Methods) we repeated PCR reaction and transformation three times and performed mini-preparations of DNA from 45 picked colonies. Only one of these contained the desired mutation and was otherwise without additional mutations. The whole procedure took one and a half months. However, even from this positive colony only low yield midi plasmid preparations could be obtained. These difficulties are described in (25) and are well known in the scientific community. To overcome these problems, we modified the codons of the SCN1A cDNA using the highly homologous human cardiac SCN5A sequence as a template, since plasmids containing SCN5A are known to be easily propagated in bacteria and efficiently expressed in heterologous systems (25).
Taking advantage of the fact that most amino acids can be coded by more than one nucleotide triplet, we devised the following strategy of redefining the cDNA sequence of SCN1A. First, SCN1A and SCN5A amino acid sequences were aligned using a standard alignment program. Next, for each position in which the same amino acid was present in SCN1A and SCN5A, the corresponding codon of the SCN1A cDNA sequence was substituted by the codon used in SCN5A. For all other amino acids, the original codon present in SCN1A was replaced by the synonymous codon being most similar to the corresponding SCN5A codon, if any. The procedure is illustrated in Figure 2.
Strategy of cDNA optimization. A short stretch of the alignment of SCN5A with SCN1A amino-acid sequences is shown together with the respective cDNA sequences. The optimized cDNA sequence is shown below with altered bases highlighted in red. For example, the codon of F1526 is changed from the original ttc to ttt, which is more similar to the codon att encoding the corresponding isoleucine in SCN5A.
Accomplishing these steps, we generated an in silico sequence in which the identity between SCN1A (length: 5994 bp) and SCN5A (length: 6048 bp) cDNAs increased from 62% to 77%. In total, 1218 positions were modified in the SCN1A cDNA, giving rise to a modified sequence that differs from the original by 20.1%. A total of 1108 codons were changed, 713 of which are coding for the same amino acid in SCN1A and in SCN5A (see Supplementary information). The modified sequence was synthesized and sub-cloned into the pcDNA3.1 vector, giving rise to a plasmid that is extremely easy to handle: It could be easily transformed in standard bacteria strains and amplified with high yield (∼100 µg from a 50 ml culture compared to ∼5 µg for the original plasmid) growing overnight at 37℃. In addition, restriction-free mutagenesis was straightforward and highly efficient, enabling the insertion of different mutations with high fidelity in the SCN1A background. Thus, from now on, we will refer to the modified sequence as the “optimized sequence” to distinguish it from the original one.
Even though the optimized sequence did not change the encoded amino acid sequence, effects on protein translation and folding cannot be excluded (31). For example, a codon modified sequence of the HERG potassium channel led to decreased translation and improved trafficking (32). Even functional properties could in principle be altered as a consequence, for example of a different folding path. We therefore first compared currents induced by the original plasmid and the optimized SCN1A plasmid. Transient transfection of both plasmids led to typical inward sodium currents in whole cell patch clamp recordings (Figure 3(a), (b)), with typical activation and inactivation curves (Figure 3(c)). Overall current density was similar in these control transfections (Figure 3(d)). Current density obtained with the original plasmid was about fourfold smaller than described earlier (12). However, absolute expression levels depend on many factors including the cell type used and the transfection procedure. Actually, the current levels obtained in these experiments allow for reliable voltage control, difficult to achieve with larger expression. More importantly, biophysical properties seem to be indistinguishable between the two plasmids, as indicated by almost identical voltages of half-maximal activation (Figure 3(e)) and half-maximal inactivation (Figure 3(f)).
Comparison of sodium currents generated by the original and by the optimized SCN1A plasmid in HEK cells. (a), (b) Representative whole-cell Na+ currents recorded from HEK cells transiently transfected with the original plasmid (a) or with the optimized plasmid (b). (c) Curves of voltage dependence of activation (squares) and of fast inactivation (triangles) relative to the experiments shown in (a) and (b). Black symbols are for the original plasmid, gray for the optimized. The small difference in the midpoint of the activation curves lies within experimental variability. (d) Peak current densities recorded from cells expressing the original plasmid (black, n = 15), or the optimized plasmid (gray, n = 11), from a set of two parallel transfections. (e), (f) Average values of voltages of half-maximal activation (e) and half-maximal inactivation (f) obtained for the original and optimized plasmid. For the original plasmid V1/2 act = −27.0 ± 4.4 mV, zact = 4.2 ± 0.8; V1/2 inact = −61.0 ± 4.3 mV, kinact = 5.8 ± 0.5 mV (n = 8). For the optimized plasmid V1/2 act = −25.9 ± 3.3 mV, zact = 4.2 ± 0.4; V1/2 inact = −61.0 ± 2.0 mV, kinact = 5.4 ± 0.6 mV (n = 7). Error bars in all panels are SD. Differences were non-significant (p > 0.2, Student’s unpaired t test).
We thus conclude that the optimized plasmid can be reliably used to determine possible biophysical effects induced by migraine-causing mutations.
Characterization of mutant Nav1.1 currents
As a further test of the validity of employing the optimized plasmid to investigate the biophysical effects of disease-causing mutations, we introduced the already described mutant L1649Q (5,23,24). In agreement with earlier work (23,24), we observed a significant reduction in surface expression (Supplementary Figure 1(a), (b)). Also, the functional properties of the mutant, characterized by an almost unaltered activation curve (Supplementary Figure 1(a), (b)), a shift of the steady state inactivation to more positive voltages (Supplementary Figure 1(d)), an acceleration of the recovery from inactivation (Supplementary Figure 1(e)), and a significant slowing of the inactivation time course (Supplementary Figure 1(f)), are in agreement with earlier studies (24) and with results obtained for the equivalent mutation in the context of the cardiac Na+ channel (5).
We next introduced in the optimized plasmid two recently discovered but functionally uncharacterized migraine mutations, L1670W (26) and F1774S (22), using a standard protocol of restriction-free (RF) mutagenesis. Positions of these mutations refer to the reference sequence of SCN1A, which is used in the genetics literature; the actual mutations in our plasmid are L1659W and F1763F, respectively. One positive clone for each mutation was amplified and fully sequenced. No recombination or spurious mutations were found.
WT and mutant channels were characterized using standard electrophysiological protocols. Both mutants expressed typical inward Na+ currents (Figure 4(a)) and activated with the same voltage dependence as WT channels (Figure 4(b); Supplementary Figure 2), with currents starting to activate at ∼−40 mV and peaking around −10 mV. However, while F1774S expressed currents of the same amplitude as WT (Figure 4(c)), the L1670W mutant exhibited a significantly smaller current density, probably due to a lower expression of the channel on the cell surface (Figure 4(c), Supplementary Figure 3). Non-stationary noise analysis was performed to distinguish whether the lower current density of the L1670W mutant was due to a smaller single channel current amplitude or to a reduction of the open probability (Supplementary Figure 4). Both parameters were essentially identical to those of WT SCN1A (Supplementary Figure 5). Thus, at least in the in vitro HEK expression system, the L1670W mutant appears to be associated with a reduced surface expression level.
Functional expression of WT and two FHM3 mutants in the optimized SCN1A plasmid in HEK cells. (a) Representative whole-cell Na+ currents recorded from HEK cells transiently transfected with WT, L1670W or F1774S Nav1.1 cDNAs. (b) Normalized current density voltage relationship determined from cells expressing WT (black circles, n = 11), L1670W (gray squares, n = 13) or F1774S (blue diamonds, n = 12) channels. For this panel, only cells with negligible series resistance error and sizable current density (>∼13 pA/pF) were used. Differences between WT and mutant are not significant (p > 0.05 for all points). (c) Peak current densities recorded from cells expressing WT (black, n = 46), L1670W (gray, n = 61) or F1774S (blue, n = 26). L1670W mutant exhibited significantly reduced current density compared to WT and F1774S (p < 0.001). Error bars indicate SEM. For this panel, cells with suboptimal voltage clamp and small current densities were also included.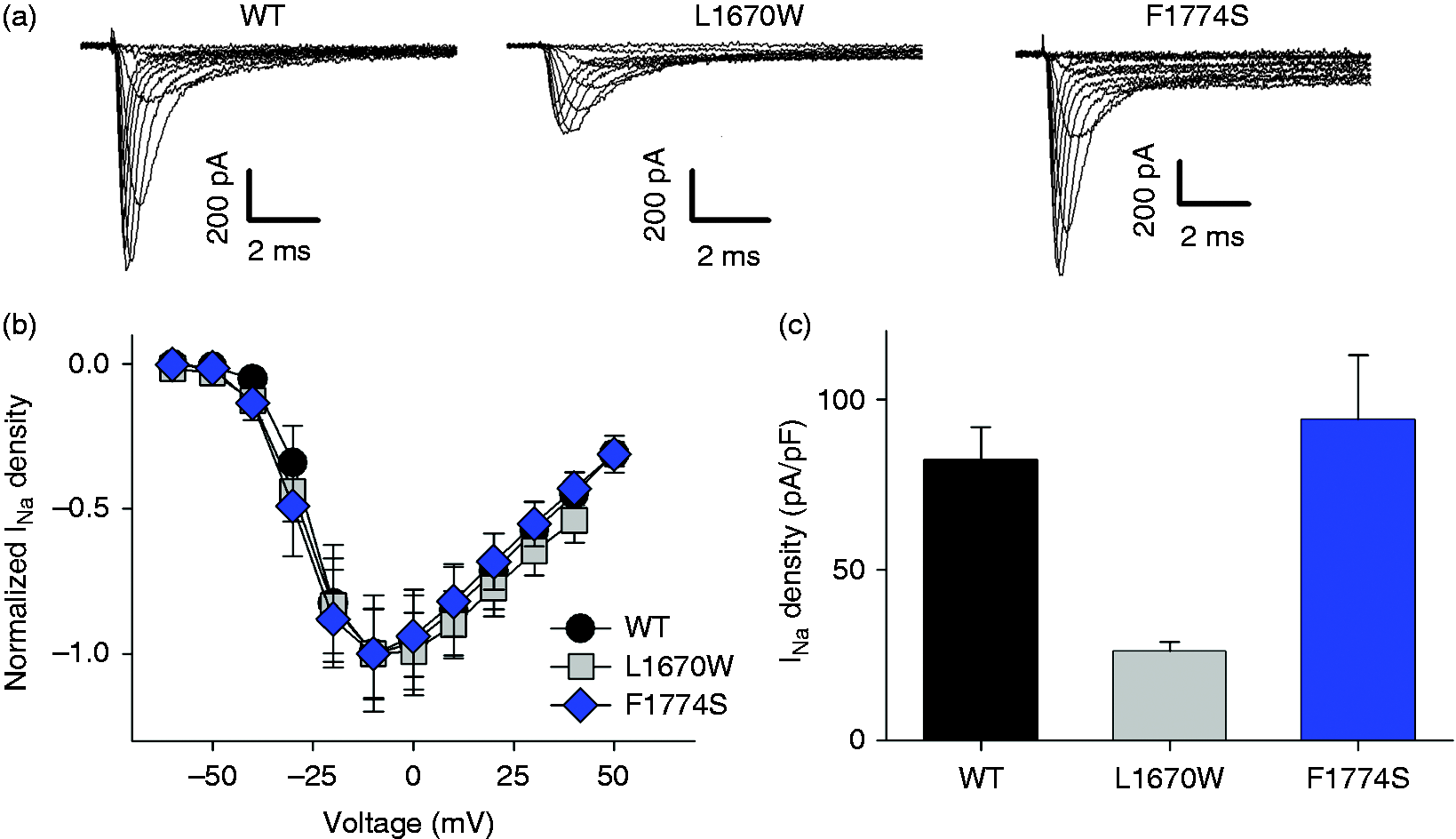
We then compared the inactivation properties of L1670W, F1774S and WT Na+ currents. The kinetics of fast inactivation, quantified by fitting a double exponential equation to the decaying current phase, was similar for both mutants and WT (Supplementary Figure 6). However, for both mutations, incomplete fast inactivation led to an increase of the persistent current measurable 70 ms after the voltage step (Figure 5(a), (b)). In addition, the incomplete inactivation allowed the detection of the time course of a very slow component of inactivation as illustrated in Figure 5(a). This slow component most likely reflects the classical “slow inactivation” process of the sodium channel. The kinetics of slow inactivation is in the hundreds of milliseconds range (33), consistent with the kinetics seen in Figure 5(a). Even after 1 second at 0 mV, the current decay is incomplete.
L1670W and F1774S mutations interfere with the inactivation process. (a) Representative current traces recorded applying a 1 s long pulse to 0 mV normalized to the absolute value of the peak current. Inset shows the initial current phase at higher time resolution. (b) Relative persistent current measured at the end of the 70 ms pulse and normalized to the absolute value of the peak current. Inset shows representative current traces recorded at 15 mV (WT: black, n = 12; L1670W: gray, n = 14; F1774S: blue, n = 14; p < 0.001 for all voltages). (c) Voltage dependence of steady state inactivation (WT: n = 14, L1670W: n = 9, F1774S; n = 13). The solid lines represent Boltzmann fits to averaged data. (d) Averaged V1/2 of steady state inactivation (p < 0.05 and p < 0.001 respectively for L1670W and F1774S mutant compared to WT). The steepness of the steady state inactivation curve was not affected by either mutation (data not shown). (e) Representative time courses of recovery from inactivation at −120 mV superimposed with double exponential fits (lines). (f) Averaged τfast of the recovery from inactivation. The time constant is significantly reduced in both mutants (WT: black, n = 12; L1670W: gray, n = 14; F1774S: blue, n = 14; p < 0.001 and p < 0.05 respectively for L1670W and F1774S mutant compared to WT).
Using the relative persistent current measured after 70 ms (Figure 5(b)), the normalized current-voltage relationship (Figure 4(b)), and the absolute current density (Figure 4(c)), we calculated the absolute persistent current density for WT and the two mutants (Supplementary Figure 7). Despite the significantly smaller current density of mutant L1670W, the absolute persistent current density is larger compared to WT (Supplementary Figure 7). In agreement with an effect of the mutations on fast inactivation, a depolarizing shift of the voltage dependence of steady-state inactivation was evident for both mutants and particularly pronounced for F1774S (WT: V1/2 = −57.2 ± 1.7 mV; L1670W: V1/2 = −49.9 ± 1.7 mV; F1774S: V1/2 =−45 ± 1.3 mV) (Figure 5(c), (d)).
To quantify the kinetics of recovery from fast inactivation, we applied a protocol in which cells were depolarized to 0 mV for 100 ms to fully inactivate the Na+ channels, and then repolarized to −120 mV with pulses of increasing duration. Peak currents at the final test pulse were analyzed by fitting a double exponential function (see Materials and Methods).
Both mutants exhibited an acceleration of the recovery manifested by a decrease of the fast time constant (τfast), particularly evident for L1670W (Figure 5(f)) while the slow time constant, τslow, and the relative area of the slow component, Arel, were similar between WT and mutant channels (Supplementary Figure 8).
To test the functional effects of the mutations in a situation of high-frequency electrical activity, as occurring, for example, in fast spiking parvalbumin positive basket cells (34), we applied a stimulation train of 200 pulses to −10 mV of 5 ms duration followed by a 3 ms period at −90 mV (corresponding to a frequency of 125 Hz) (Figure 6). In WT, a large drop of current was observed after the first pulse (see the first spikes in Figure 6(a), (b)) and subsequent peak current values decayed with a time course that could be well fitted by a single exponential function. Conversely, both mutant channels exhibited a smaller initial drop after the first pulse and peak currents decayed significantly less during the train (Figure 6(a), (b)). The decay constants for mutations L1670W and F1774S were more than double compared to WT (Figure 6(c)). The faster recovery from inactivation of the mutant channels might explain the decreased accumulation of inactivation during high frequency stimulation.
Accumulation of inactivation during high frequency stimulation. (a) Representative traces of the response to a stimulation train of 200 pulses to −10 mV of 5 ms duration and a 3 ms period at −90 mV between the pulses. (b) Peak current decay of the traces shown in (a) as a function of pulse number. (c) Decay constants obtained from single exponential fits to data as those shown in panel B (WT: n = 12, L1670W: n = 7, F1774S: n = 16; p < 0.001 for both mutants).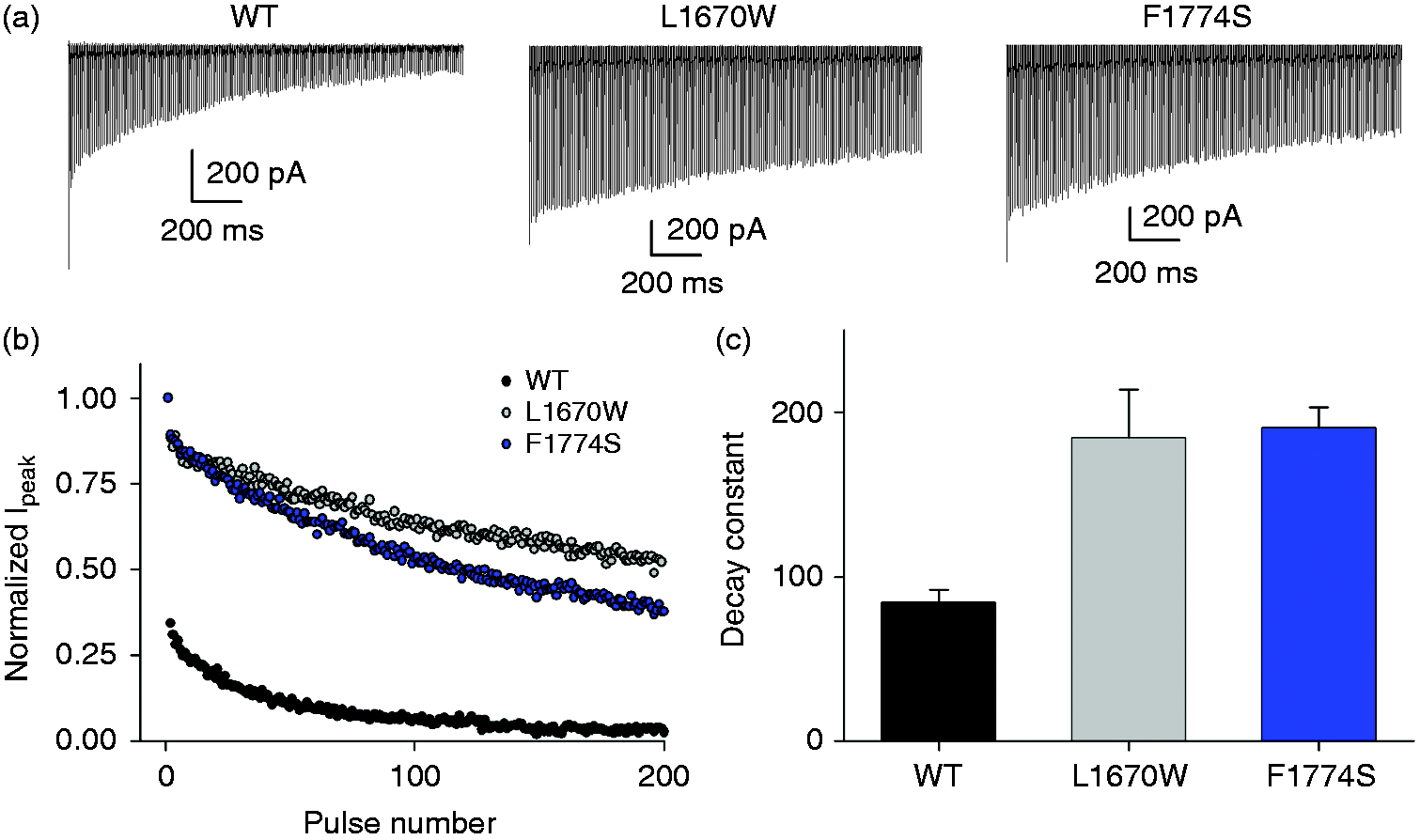
Discussion
In summary, in the present work we developed an optimized SCN1A cDNA plasmid that overcomes technical difficulties in bacterial amplification and mutagenesis and used the plasmid to characterize two SCN1A mutations associated with hemiplegic migraine. Mutant L1670W was reported to co-segregate with pure FHM3 in a Chinese family in which all affected members displayed very similar migraine attacks (26). Similarly, the de novo F1774S mutation was identified in a female patient manifesting classical symptoms of pure hemiplegic migraine without signs of epilepsy in EEG recordings (22). Both mutations induce modifications in gating properties that are consistent with a gain of function of Nav1.1 underlying the disease. Mutant F1774S, as the recently characterized L1624P (13), displays a pure gating effect. In contrast, L1670W shows a significant reduction of the current density in transfected HEK cells. As the single channel current amplitude and the peak open probability of this mutant are unaltered, the reduced current density likely reflects a folding defect leading to a smaller number of channels expressed in the plasma membrane, as found in an even more drastic manner for the L1649Q mutant (12,23). It remains to be discovered if the reduced membrane insertion reflects an artifact of the HEK cell expression system or if also in vivo the mutation induces a folding defect. If this is the case, it will be important to find out which of the two major effects, the alteration of gating properties or the reduction of membrane expression, is the most important for the migraine phenotype.
The increase of persistent currents seen for both mutants is dramatic. While in WT they amount to less than 1.5% of the peak current, in L1670W and F1774S the percentage increases more than sixfold, being in the order of 10%. The persistent currents decay very slowly, with a time constant in the seconds time range (see Figure 5(b)), most likely reflecting the process of “slow inactivation” (33). A destabilization of fast inactivation is also highlighted by the depolarizing shift of steady state inactivation and by the faster recovery from inactivation. All these functional effects are expected to have a large effect on the neuronal firing properties. Indeed, the accumulation of inactivation during a high frequency train of stimulation is markedly reduced for both mutants.
Thus, our results are in line with the general idea of an overall gain of function of the Nav.1.1 channel as a possible explanation of the cause of FHM3. The final outcome of such a gain of function in the context of the in vivo neural network, and how it triggers or increases the susceptibility to migraine attacks, is difficult to predict, also because the relative contribution of the Nav1.1 channel to the overall Na+ current of various neuronal cell types is largely unknown.
From a biophysical point of view, it is interesting to map the migraine mutations onto the recently determined 3D structures of voltage-gated Na+ channels (Figure 1). L1670 is located in a region where the S4-S5 linker of domain IV, folded as an alpha helix in the structures of both the cockroach and the electric eel Na+ channel (28,35), bends into the S5 transmembrane helix. Activation of the voltage sensor of domain IV has been implicated in fast inactivation (36). The fact that inserting a bulky tryptophan residue in this S4-S5 region strongly affects inactivation is in agreement with the hypothesis that the initial part of S5 is involved in the conformational changes following DIV voltage sensor outward movement. F1774 is located in the center of S6 of domain IV (Figure 1). Interestingly, in the cockroach structure this part of S6 is in direct contact with the C-terminal end of the first pore helix of domain IV. It can be speculated that this region is involved in interacting with the putative inactivation particle that is assumed to enter and block the pore once channels are opened (36). Interestingly, in alanine scanning mutagenesis studies of the S4-S5 loop and of the S6 segment of rat Nav1.2, the residues corresponding to L1670 and F1774, respectively, emerged among those with a major effect on fast inactivation and an increase of the persistent current (37,38), confirming again that hemiplegic migraine is associated with a defective inactivation process.
The results obtained for the two FHM3 mutations validate our procedure of cDNA sequence refining to overcome the known technical challenges associated with SCN1A bacterial amplification and mutagenesis. For the optimization, we used the sequence of the highly homologous human SCN5A as a template as this variant does not manifest particular difficulties in plasmid handling. Optimization resulted in the introduction of a large number (1218) of nucleotide changes. This cannot be considered a priori non-influential at the functional level, and potential risks associated with this approach have to be discussed. First, even though, due to the genetic code degeneracy, a protein can be synthesized based on multiple synonymous mRNA sequences, it has been well established that codons are used in a non-random manner, with organism-specific codon preferences (39). The codon usage plays relevant roles in determining mRNA editing, mRNA stability, speed of protein translation, accuracy of protein folding, and level of protein expression and secretion (39,40). Thus, when a sequence optimization is performed, it is crucial to check the suitability of the new construct. Several algorithms are available to test for the presence of rare codons compared to the general codon usage in a given organism (41). A frequently used global parameter characterizing a given sequence is the Codon Adaptation Index (CAI), which compares the codon usage of a sequence to the codon usage of a set of highly expressed genes (39). The CAI of human SCN1A (0.756) is considerably smaller than that of the cardiac SCN5A isoform (0.846) (Supplementary Table 1), while the value of the optimized plasmid lies in between (0.816, Supplementary Table 1), indicating a higher usage of frequent codons compared to the original sequence. A finer, sequence-dependent measure of codon usage is given by the %MinMax score that is calculated over a sliding window (39,41) (Supplementary Figure 9). It can be seen that our optimization strategy to “convert” as far as possible the SCN1A sequence towards that of SCN5A significantly reduces the percentage of rare codon clusters disseminated in the SCN1A sequence (Supplementary Figure 9). The high percentage of rare codons in the SCN1A sequence is rather surprising. It may be speculated that rare codons result in a slowing of mRNA translation, which might help in proper folding of such a large membrane protein. However, the cardiac Na+ channel is equally complex at the protein level but exhibits only a small percentage of rare codons. More generally, while effects of codon usage are likely relevant, at the moment a specific predictive correlation between the quantity or quality of usage of synonymous codons and the impact on protein biogenesis is not possible. Thus, even though we cannot exclude that several aspects of the biogenesis of the “optimized” Nav1.1 protein are altered, the only way to test possible effects is provided by biochemical and functional analysis. Our experimental results are encouraging, in that the overall expression level of the optimized plasmid is comparable to that of the original clone, suggesting a similar efficiency of protein folding. Even more importantly, the electrophysiological data show that the functional biophysical properties of the native and optimized channel are practically identical.
Conclusions
We expect that the optimized plasmid will be useful for the investigation of a broad spectrum of epilepsy-associated SCN1A mutations in heterologous systems. More generally, a similar sequence optimization can be likely applied also to other Nav encoding cDNAs, thus enlarging the panorama of neurological and muscular diseases whose study can be facilitated by the application of this strategy.
Article highlights
A SCN1A-encoding optimized plasmid has been designed and synthesized. SCN1A-plasmid amplification and mutagenesis are highly improved by optimization. FHM3-causing mutations impair the Nav1.1 current inactivation mechanism. FHM3 associated Nav1.1 currents show a gain of function phenotype.
Supplemental Material
Supplemental material alignment -Supplemental material for Gain of function of sporadic/familial hemiplegic migraine-causing SCN1A mutations: Use of an optimized cDNA
Supplemental material, Supplemental material alignment for Gain of function of sporadic/familial hemiplegic migraine-causing SCN1A mutations: Use of an optimized cDNA by Sara Bertelli, Raffaella Barbieri, Michael Pusch and Paola Gavazzo in Cephalalgia
Supplemental Material
Supplemental material for Gain of function of sporadic/familial hemiplegic migraine-causing SCN1A mutations: Use of an optimized cDNA
Supplemental material for Gain of function of sporadic/familial hemiplegic migraine-causing SCN1A mutations: Use of an optimized cDNA by Sara Bertelli, Raffaella Barbieri, Michael Pusch and Paola Gavazzo in Cephalalgia
Footnotes
Acknowledgments
We thank Dr. Massimo Mantegazza for providing us with the original SCN1A expression construct and Francesca Quartino for excellent technical assistance.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was partially supported by a grant from the Telethon Foundation (GGP17178 to P Gavazzo). Sara Bertelli is supported by the PhD program of SISSA.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.