
Abstract
Background
The biological mechanisms of headache chronification are poorly understood. We aimed to identify changes in DNA methylation associated with the transformation from episodic to chronic headache.
Methods
Participants were recruited from the population-based Norwegian HUNT Study. Thirty-six female headache patients who transformed from episodic to chronic headache between baseline and follow-up 11 years later were matched against 35 controls with episodic headache. DNA methylation was quantified at 485,000 CpG sites, and changes in methylation level at these sites were compared between cases and controls by linear regression analysis. Data were analyzed in two stages (Stages 1 and 2) and in a combined meta-analysis.
Results
None of the top 20 CpG sites identified in Stage 1 replicated in Stage 2 after multiple testing correction. In the combined meta-analysis the strongest associated CpG sites were related to SH2D5 and NPTX2, two brain-expressed genes involved in the regulation of synaptic plasticity. Functional enrichment analysis pointed to processes including calcium ion binding and estrogen receptor pathways.
Conclusion
In this first genome-wide study of DNA methylation in headache chronification several potentially implicated loci and processes were identified. The study exemplifies the use of prospectively collected population cohorts to search for epigenetic mechanisms of disease.
Introduction
In a number of patients who suffer from episodic headache attacks, their headache transforms into a chronic form (1,2). Chronic headache, defined as headache occurring on at least 15 days per month, affects 3–5% of the general population and 40% of patients in headache clinics, and is associated with severe disability and impaired quality of life (1,3). Approximately half of these patients have chronic migraine (1). Established risk factors for headache chronification include the overuse of acute headache medications and a high baseline attack frequency (4). The biological mechanisms involved in the transition from episodic to chronic headache are, however, unknown.
In recent years it has become evident that epigenetic processes play an important role in a range of multifactorial diseases (5). Epigenetics entail changes in gene expression that are not due to variation in the DNA sequence. A main epigenetic mechanism is DNA methylation, the covalent addition of a methyl group to the 5th carbon of cytosine residues, which is typically associated with gene silencing (5,6). Epigenetic modifications can be stably inherited through cell divisions but can also be dynamic. They enable cell and developmental stage-specific regulation of gene expression, and play an important role in programming lasting responses to environmental cues (6,7). Due to the ability of DNA methylation changes to shift a biological system from one stable state to another, their implication in headache chronification is of particular interest. It has been hypothesized that frequent headache attacks may lower the threshold for subsequent headache attacks through epigenetic mechanisms resulting in a feed-forward loop (6). Similarly, psychological stress and female sex hormones, which have been implicated in migraine genesis, are known to exert their physiological effects partly through epigenetic mechanisms (6,8).
In this study, subjects were recruited from the Norwegian population-based Nord-Trøndelag Health (HUNT) Study, where they gave blood samples and were assessed for headache at two time points 11 years apart. In order to identify changes in DNA methylation associated with headache chronification we compared cases who transformed from episodic to chronic headache during the follow-up period with controls who did not develop chronic headache. For all selected subjects, genome-wide DNA methylation levels were characterized at baseline and follow-up.
Materials and methods
Participants
In the HUNT2 (1995–1997) and HUNT3 (2006–2008) studies, the entire population of the Norwegian county of Nord-Trøndelag, 20 years of age or older, was invited to complete extensive questionnaires concerning health-related issues, participate in a clinical examination and donate blood samples. Among 92,566 individuals in HUNT2 and 94,194 in HUNT3 invited to participate, 51,383 (56%) and 39,701 (42%), respectively, answered the headache questionnaire (9,10).
The headache classification used in this study has been described in detail elsewhere (9–11). Briefly, in each survey those who answered ‘yes’ to the screening question ‘Have you suffered from headache during the last year?’ were defined as headache sufferers and asked to fill in the other headache questions. Participants indicated the number of days with headache per month, with the options <7, 7–14 or >14 days/month in HUNT2 and the options <1, 1–6, 7–14 or >14 days/month in HUNT3. Chronic daily headache (CDH) was defined as headache occurring on ≥15 days/month. In HUNT2, acute medication overuse was defined as ‘analgesic use daily or almost daily for ≥1 month during the last 12 months’, and in HUNT3 as ‘over-the-counter-medication use for headache ≥4 days/week during the last month’. Medication-overuse headache was defined as the combination of CDH and acute medication overuse (9,11,12). This headache classification has been validated by interview diagnoses (9,10). In HUNT2, the sensitivity and specificity were, respectively 85% and 83% for headache suffering (Cohen’s kappa κ = 0.57), and 91% and 56% for headache frequency <7 days per month (κ = 0.50). In HUNT3, the sensitivity and specificity were, respectively 88% and 86% for headache suffering (κ = 0.70), 69% and 99% for CDH (κ = 0.75) and 75% and 100% for medication-overuse headache (κ = 0.75).
Cases for the current study were selected among participating women who at the time of HUNT2 (baseline) were ≤35 years of age and reported headache on <7 days/month, and at the time of HUNT3 (follow-up) reported ≥15 headache days/month (CDH). To avoid sample heterogeneity resulting from different DNA methylation patterns in men and women, only women were included. Likewise, to avoid DNA methylation changes resulting from the onset of menopause during follow-up only women ≤35 years of age at baseline were included. In total 43 subjects fulfilled these criteria. After excluding samples with missing DNA from either baseline or follow-up, 40 cases were included in the study. We selected an equal number of controls, who were matched with cases on their headache frequency and age at baseline, but at follow-up either did not have headache anymore or had headache on <7 days/month. Average time between baseline and follow-up examinations was 11.4 years (range 10–12.6 years).
Ethics
Signed informed consent was obtained from all participants in the HUNT2 and HUNT3 surveys, and the present study was approved by the Regional Committee for Medical Research Ethics (2012/2316/REK midt).
Genome-wide quantification of DNA methylation
A detailed description of sample handling, quantification of DNA methylation, data normalization and quality control is given in Supplemental Methods. Briefly, we performed array quantification of DNA methylation on genomic DNA isolated from leukocytes, after bisulfite conversion, using the Illumina Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA). For Stage 1, 96 samples from 24 cases and 24 controls were analyzed. For Stage 2, 64 samples from 16 cases and 16 controls were analyzed. The methylation level at each CpG site for each sample was calculated as a β-value, scaling from 0 (not methylated) to 1 (fully methylated). After sample-wise quality control steps, 36 cases and 35 controls contributed to the final analysis. An overview of the study design and participants in the final analysis are given in Figure 1. After marker-wise quality control steps, out of 485,775 CpG markers initially interrogated 431,282 and 436,517 markers were available for analysis in Stage 1 and Stage 2, respectively. No differences in estimated white blood cell ratios were found between patients and controls for Stage 1, nor for Stage 2 (Supplemental Tables 2 and 3, and Supplemental Methods), therefore we did not include the ratios as cofactors in the analysis.
Diagram of the study design and participants. A total of 36 female headache patients who transformed from episodic to chronic headache (‘cases’) between examinations at baseline and follow-up were matched against 35 female headache patients who did not transform to chronic headache (‘controls’).
Analytic approach
Baseline characteristics
Baseline characteristics were compared between cases and controls by Fisher’s exact test (for binary variables) or independent samples t-test (for continuous variables). The examined variables included known or suggested risk factors for the development of chronic daily headache (4,13); age (continuous); low educational level (upper secondary school or less as highest education); self-reported daily use of tranquilizers last year (yes, no); current daily smoking (yes, no); low physical activity (<1 hour light exercise per week); body-mass index (continuous); Hospital Anxiety and Depression Scale (HADS) for each of anxiety and depression (continuous) (14), chronic musculoskeletal complaints (yes, no) (15); gastrointestinal complaints (yes, no) (16), self-reported diabetes mellitus (yes, no) and self-reported rheumatoid or osteoarthritis (yes, no). Of the participants included in the study, 3 had missing information on physical activity and 1 on chronic musculoskeletal complaints.
Association analysis and replication
The effect of headache chronification (case-control status) was modeled on changes in DNA methylation levels. For each individual, normalized methylation level estimate (β-value) at baseline was subtracted from methylation level estimate at follow-up for each CpG site. Resulting methylation level changes at each CpG site were then compared between cases and controls using linear regression model adjusted for age, using the lm package in R software (17). Data from each stage was analyzed separately and the 20 CpG sites showing strongest association with chronification in Stage 1 were taken forward for replication in Stage 2. A Bonferroni-corrected p-value threshold (0.05/20 = 0.0025) was used to consider statistically significant replication.
Meta-analysis
Subsequently, a fixed-effects meta-analysis was performed on DNA methylation data from Stage 1 and Stage 2, weighted by the inverse variance, using the GWAMA software (18). In total 436,549 CpG markers were present in either stage after quality control and could be used in the analysis, resulting in a Bonferroni-corrected p-value threshold of 1.15 × 10−7 (0.05/436,549).
Characterization of top-ranking CpG sites
Functional profiling of the meta-analysis top-ranking CpG sites was carried out using g:Profiler web-based software (19), see Supplemental Methods for a full description. Briefly, g:Profiler performs a statistical gene set enrichment analysis to find functional gene groups or biological pathways that are significantly over-represented in user-provided input. The results presented are based on the 200 top-ranking CpG sites from the meta-analysis, and results were considered significant if the multiple testing corrected p-value for the enriched group or pathway was ≤.05. Characterization of gene expression in various human tissues was retrieved from the GTEx database (20). Although DNA methylation is largely tissue-specific, inter-individual variation in DNA methylation has been shown to correlate between blood and brain for a subset of CpG sites (21). Correlation of DNA methylation levels between blood and brain at the top CpG sites was analyzed using the Blood Brain DNA Methylation Comparison tool (http://epigenetics.essex.ac.uk/bloodbrain) that utilizes DNA methylation data of blood and four different brain regions from matched samples (21).
Analysis of medication overuse
Overuse of acute headache medications is a known risk factor for headache chronification. To evaluate to what extent medication overuse influenced the identified associations, the association analysis and meta-analysis was repeated stratified by medication overuse. Cases with or without medication overuse at follow-up were analyzed separately against all controls. Results were visualized as forest plots using the metafor R package (version 1.9-8) (22). Additionally, unsupervised clustering of the samples was performed using the hclust R package, based on their methylation levels at follow-up at each of the 200 top-ranking CpG sites from the initial meta-analysis.
Results
Demographic and clinical characteristics at baseline for cases and controls.

n.a. = not applicable.
Independent samples t-test. bFisher’s exact test.
Summary statistics of the 20 CpG sites that showed strongest association with headache chronification in Stage 1.

β, SE (standard error) and p (p-value) for association between headache chronification and change in CpG site methylation (linear regression model), adjusted for age. Chr = chromosome. Position = genome position (build GRCh38). *For probes not located in direct relation to a gene, the two nearest genes are listed, separated by semicolon.
CpG loci demonstrating strongest association to headache chronification in combined meta-analysis.

Methylation levels in blood showing high correlation with methylation levels in one or more regions of the human brain at p-value < .05. **For CpG markers not located in direct relation to a gene, the two nearest genes are listed. β, SE (standard error) and p (p-value) for association between headache chronification and change in CpG site methylation in meta-analysis of results from Stages 1 and 2. Chr = chromosome. Position = genome position (build GRCh38).

Correlation of DNA methylation in blood and four brain regions for cg01010870 (SH2D5). Top: Boxplot of the distribution of DNA methylation values for cg01010870 split by tissue. Bottom: Four scatterplots demonstrating the relationship between DNA methylation in whole blood and four brain regions: PFC = pre-frontal cortex; EC = entorhinal cortex; STG = superior temporal gyrus; CER = cerebellum. Plots are retrieved using the Blood Brain DNA Methylation Comparison tool (21).
Functional profiling of top associated CpG sites and corresponding genes.

Functional profiling using g:Profiler. The p-values are corrected for multiple testing. Highest-level hierarchical groups and pathways (‘main terms’) that were significant are marked in bold. Increased indent indicates deeper level in the local hierarchy under the group or pathway.
When stratifying the meta-analysis for overuse of acute headache medications among cases, the associations for the 10 top-ranking CpG sites from the initial meta-analysis had the same direction and were of comparable strength in each of the two groups (Figure 3). Likewise, unbiased hierarchical cluster analysis based on the 200 top-ranking CpG sites did not discriminate between cases with and without medication overuse (Figure 4).
Forest plots showing meta-analysis results separately for cases with and without medication overuse. Meta-analysis beta-value (Beta) and its 95% confidence intervals (95% CI) for the top 10 CpG sites (cg01010870 to cg14118171) from the meta-analysis. Unsupervised clustering of cases based on the top 200 CpG sites from the meta-analysis. Unsupervised hierarchical clustering using methylation status at follow-up did not discriminate between cases with (marked in yellow) and without medication overuse in either Stage 1 (A) or Stage 2 (B).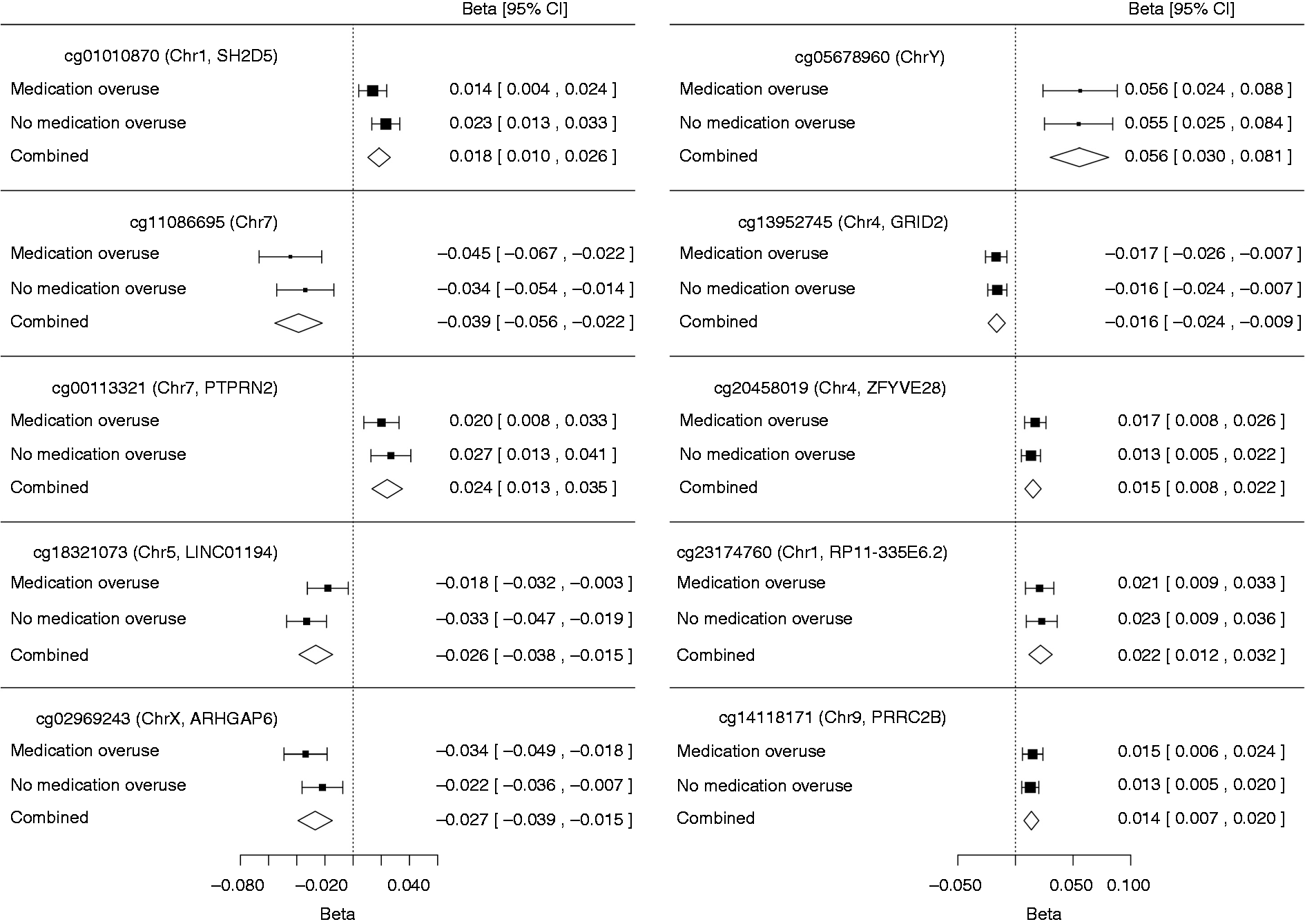

Discussion
This is, to our knowledge, the first study of epigenetic changes in headache chronification. Using a retrospective case-control design, DNA methylation changes were examined across the genome comparing patients who converted from episodic to chronic headache during an 11-year follow-up period with matched controls. In the discovery stage several CpG sites showed suggestive association to headache chronification, but these failed to replicate in the second stage. Combining association results from both stages in a fixed-effects meta-analysis we identified CpG sites that may point to genes involved in headache chronification. An enrichment was found for genes related to calcium ion binding, biological adhesion, and intracellular estrogen receptor signaling.
Since cases in this study reported episodic headache at baseline and CDH at the time of follow-up, it is likely that they represent transformed rather than de-novo chronic headache. However, a limitation is that the questionnaire-based assessment did not allow distinction of chronic migraine from other types of chronic headache. Previous epidemiological studies suggest that approximately half of those with CDH fulfill criteria for chronic migraine (1), making it likely that migraineurs contribute substantially to our case group.
Medication overuse is a major risk factor for headache chronification, and it has previously been shown that medication overuse headache accounts for about half of the participants who report CDH in the HUNT population (13). A similar proportion was seen in this study with 17 of 36 cases (47%), but none of the controls, reporting medication overuse at the time of follow-up. Notably, all 4 participants who reported medication overuse at baseline had developed chronic headache at follow-up. Given our design, it is feasible that at least some of the DNA methylation changes result from medication use. Still, when stratifying the meta-analysis results by cases with and without medication overuse, the strength and direction of association for the top 10 loci from the meta-analysis were similar between the groups (Figure 3). Also, unsupervised hierarchical clustering of the samples based on methylation levels for the top 200 CpG sites did not discriminate cases with from cases without medication overuse (Figure 4). The results therefore suggest that medication overuse is unlikely to explain the identified associations.
The most significant CpG site is at the SH2D5 gene, which encodes the SH2 Domain Containing 5 protein. The second strongest associated CpG site is 76 kb downstream from the nearest gene NPTX2, which encodes the Neuronal Pentraxin II protein. Both proteins are highly expressed in the adult human brain (23,24) (Supplemental Figure 1), and DNA methylation levels at the associated CpG sites are highly correlated between brain and blood (Table 3 and Figure 2). Both proteins have presumed roles in regulating synaptic plasticity. The SH2 Domain Containing 5 protein regulates synaptic plasticity through the control of Rac-GTP levels (25). Neuronal Pentraxin II regulates synaptic plasticity by acting as an inhibitor of excitatory synapses, through binding and clustering of glutamatergic AMPA receptors. Neuronal Pentraxin II is induced by neuronal activity, and has an assumed function in rebalancing excitation-inhibition after periods of increased synaptic activity (23). Of interest, another of the top associated CpG markers is related to glutamate signaling. GRID2 encodes the glutamate receptor δ2, an ionotropic glutamate receptor which is expressed in several brain regions, with highest expression in the cerebellum (20,26). Epigenetic alteration of synaptic plasticity has been previously hypothesized as a possible mechanism of migraine chronification (6). Support for this comes from the experimental finding that synchronous neuronal activity, which also occurs in cortical spreading depression of the migraine aura (27), results in DNA methylation changes in brain-specific genes related to neuronal plasticity (28). One may envisage that as neuronal activity can cause epigenetic changes altering synaptic plasticity, migraine attacks in a feed-forward loop may promote subsequent migraine attacks leading to chronification. Our findings seem to provide at least some support for a role of synaptic plasticity in headache chronification. Regardless, it is far from clear whether such changes are a cause or consequence of frequent headache attacks. Furthermore, our findings may implicate that glutamatergic activity is implicated not only in migraine pathogenesis (29), but also in the transition from episodic to chronic headache.
When considering the more extensive list of 200 top-ranking CpG sites, there is a statistically significant enrichment in genes related to several specific functional groups and pathways. The strongest enrichment was seen for calcium ion binding. Calcium ion binding and signaling is important in a wide range of cellular systems, including the synaptic release of neurotransmitters (30). While this presents a plausible mechanism for calcium ion imbalance in migraine pathogenesis, no genetic association to this or other calcium channels have been found for the common forms of migraine (31). The second strongest enrichment was found for genes involved in biological cell adhesion. Cell adhesion molecules have multiple roles in tissue development and function. In the human brain they play a major role at synapses, where they control synaptic morphology and plasticity, and at the neurovascular unit they form the molecular interface between the vascular and the central nervous system and are involved in the complex signaling between these systems (32,33). Lastly, in this study of female subjects, we found enrichment for genes involved in estrogen receptor signaling. This corresponds well with lines of evidence suggesting a role of estrogen in migraine pathogenesis (34). Estrogen receptor signaling has also been discussed specifically in relation to possible epigenetic mechanisms in migraine, since the estrogen receptors predominantly assert their action through the epigenetic programming of target genes (6).
Strengths of this study include the genome-wide assessment of DNA methylation sites, which allowed us to search for genetic loci associated with headache chronification without relying on previous hypotheses. Also, the headache classification used in this study has been validated by clinical interview. Given the longitudinal design we could determine DNA methylation levels in both cases and controls at two time points, before and after the development of chronic headache, which gave the possibility to detect changes in DNA methylation levels occurring during the natural course of the disease.
Our study has clear limitations. Despite a large baseline population of 51,000 individuals with available DNA who were characterized with respect to headache, and a follow-up time of 11 years, only 36 cases fulfilled inclusion criteria and could be used in the final analysis. This resulted in limited statistical power, as indicated by the failure to replicate signals identified in the discovery stage in the smaller replication sample. We therefore focused our subsequent analyses on the combined dataset of both stages in order to maximize the power to detect true signals. The loci identified from the combined analysis aggregate in functionally meaningful pathways, indicating the presence of true signals. Nevertheless, the reported associations will require replication in independent samples, but given our complex design this will be challenging. This clearly demonstrates the difficulty of designing studies of epigenetic changes during the natural course of incident disease, and suggests that international collaboration is required to achieve robust sample sizes when using such a study design. Another limitation was that we determined DNA methylation in leucocytes from whole blood, whereas DNA methylation patterns are to a considerable extent tissue-specific, so our findings may not be representative of changes occurring in the more relevant tissue in headache, in particular the brain (8). However, for the majority of the top associated markers, DNA methylation levels in blood correlated well with DNA methylation levels in one or more brain regions, suggesting that our design has good potential to detect DNA methylation relevant to migraine. This is also in line with previous studies that have found that DNA methylation patterns in non-affected tissues such as blood can be of value, even leading to treatment decisions (35).
A better understanding of the mechanisms underlying headache chronification will be important for developing better strategies to treat and prevent headaches in these severely affected patients. Epigenetic changes represent modifiable factors, and thereby offer potential targets for novel drug therapies (5). Although candidate regions identified in this study require independent replication and further characterization, this is a first step towards understanding epigenetic changes occurring in headache chronification.
Article highlights
The two CpG sites with strongest association to headache chronification were found in relation to genes that regulate synaptic plasticity. Associated CpG sites also showed enrichment for genes involved in calcium ion binding, cell adhesion and estrogen receptor pathways. The results suggest possible mechanisms by which epigenetic DNA methylation may be involved in the chronification of headache.
Footnotes
Acknowledgments
The Nord-Trøndelag Health Study (The HUNT Study) is a collaboration between HUNT Research Centre (Faculty of Medicine, Norwegian University of Science and Technology, NTNU), Nord-Trøndelag County Council, Central Norway Health Authority, and the Norwegian Institute of Public Health. The authors would like to acknowledge Dr. Tsun-Po Yang, Dr. Eshwar Meduri and Dr. Matti Pirinen for their advice on data normalization and analysis methods.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Research Council of Norway (grant 231187/F20 to BSW.). PP was supported by the Nordic Information for Action eScience Center (NIASC); a Nordic Center of Excellence financed by NordForsk (project number 62721) and by Estonian Research Council project IUT20-60 Omics for health: an integrated approach to understand and predict human disease. The research leading to these results has received funding from the European Union’s Seventh Framework programme project EUROHEADPAIN project (grant number 602633 to AP & AMvdM) and the Center for Medical Systems Biology (CMSB) established in the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research (NGI/NWO) (project number. 050-060-409 to AMvdM).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.