
Abstract
Background
Hypoxia causes secondary headaches such as high-altitude headache (HAH) and headache due to acute mountain sickness. These secondary headaches mimic primary headaches such as migraine, which suggests a common link. We review and discuss the possible role of hypoxia in migraine and cluster headache.
Methods
This narrative review investigates the current level of knowledge on the relation of hypoxia in migraine and cluster headache based on epidemiological and experimental studies.
Findings
Epidemiological studies suggest that living in high-altitude areas increases the risk of migraine and especially migraine with aura. Human provocation models show that hypoxia provokes migraine with and without aura, whereas cluster headache has not been reliably induced by hypoxia. Possible pathophysiological mechanisms include hypoxia-induced release of nitric oxide and calcitonin gene-related peptide, cortical spreading depression and leakage of the blood-brain barrier.
Conclusion
There is a possible link between hypoxia and migraine and maybe cluster headache, but the exact mechanism is currently unknown. Provocation models of hypoxia have yielded interesting results suggesting a novel approach to study in depth the mechanism underlying hypoxia and primary headaches.
Introduction
Hypoxia causes secondary headaches associated with low oxygen tensions, such as high-altitude headache (HAH) and headache due to acute mountain sickness (AMS) (1,2). Hypoxia may also play an important role in primary headaches such as migraine and cluster headache (CH) (3–17). Interestingly, the phenotype of HAH and AMS includes migraine features (Table 1), and as many as 65% of severe AMS cases fulfill the criteria for migraine (3). Furthermore, two epidemiological studies (4,5) reported high prevalence of migraine in a high-altitude population, and sumatriptan has shown some effect in HAH and AMS (6–9). The fact that oxygen inhalation is highly effective in CH (17,18) and the fact that obstructive sleep apnea (OSA), which is accompanied by desaturation (12), is more prevalent among CH patients (13–15) also points to hypoxia being linked to the pathophysiology of CH. Yet, possible hypoxic mechanisms in primary headaches are unknown even though several mechanisms have been proposed (Figure 1). This review focuses on the possible role of hypoxia in migraine and CH and discusses possible pathophysiological mechanisms.
Potential hypoxic mechanisms in migraine and cluster headache pathophysiology. Diagnostic criteria for migraine with aura, cluster headache, high-altitude headache and acute mountain sickness. Adapted from the third edition of the International Classification of Headache Disorders, third edition beta (ICHD-III beta) (1).

All currently known pharmacological triggers of migraine are vasoactive substances (19). Headache is unlikely to be caused by mechanical dilation per se, but rather by sensitization of perivascular nociceptors (20–22). In humans, hypoxia causes an increase in cerebral blood flow (CBF) (23,24) via vasodilation of cerebral arteries (25). During hypoxia, such sensitization could occur via NO (24), and via CGRP (26) and activation of the cAMP pathway (27). Adenosine may mediate hypoxic vasodilation (28,29) and adenosine levels in the brain increase under hypoxic conditions in rats (30,31). In healthy volunteers, 80–120 µg adenosine induces mild headache and dilation of the superficial temporal artery (32). Hypoxia may induce cortical spreading depression (see text for details). Autonomic regulation may play a role in CH (see text for details).
Cerebral physiological response to hypoxia
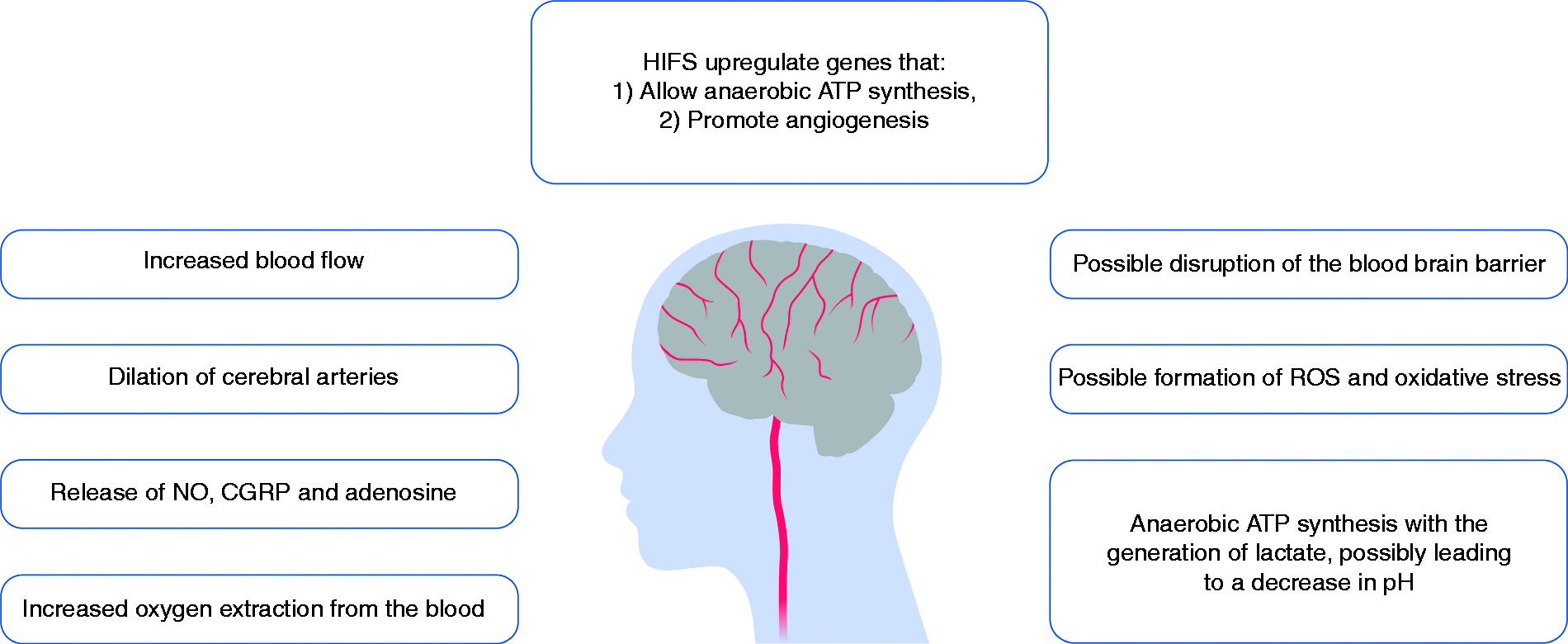
Adaptive mechanisms ensure adequate cerebral perfusion during mild to moderate hypoxia both in the short and long term (Figure 2). Peripheral and central chemoreceptors detect changes in oxygen tension, carbon dioxide tension, pH and temperature of the blood, and mediate autonomic compensatory responses (33). Therefore, hypoxemia leads to increased ventilation, sympathetic activity and blood pressure (33). Baroreceptors, cerebral autoregulation and neurovascular coupling ensure adequate cerebral blood pressure (34). Figure 3 summarizes the cerebral effects of hypoxia. As such, short-lasting reductions in atmospheric pressure do not necessarily lead to reduced oxygen saturation in the peripheral circulation and especially in the brain tissue.
Sensing hypoxia and hypothalamic coordination of the response. The cerebral effects of hypoxia.
When the arterial oxygen partial pressure (PaO2) falls below 8 kPa (= 60 mmHg), the peripheral chemosensors in the carotid and aortic bodies increase their firing rate. Glossopharyngeal afferents from the carotid bodies, and vagal afferents from the aortic bodies, relay this information centrally, to the nucleus of the solitary tract (NTS). Here they contact neurons that project to the paraventricular nucleus (PVN) of the hypothalamus, where the hypoxic response is coordinated. Neurons from the PVN project to the respiratory centers (ventral respiratory group (VRG) and dorsal respiratory group (DRG)) of the medulla and pons. Stimulation of these centers leads to increased ventilation via activation of the phrenic nerves and the diaphragm (35). Neurons from the PVN also project to the autonomic regulatory center of the medulla oblongata, the rostral ventrolateral medulla (RVLM). Here they contact neurons that project to the intermediolateral nucleus (IML) of the spinal cord and activate efferent sympathetic nerves, leading to an increased heart rate (35).
Initially, considerable reductions in arterial oxygen partial pressure can be compensated by increased oxygen extraction, due to the sigmoidal shape of the hemoglobin-oxygen dissociation curve (33). When the oxygen saturation falls below 90%, the cerebral arteries dilate (23,25) and cerebral blood flow (CBF) increases (23–25). The vasodilation could be mediated by nitric oxide (NO) (24), calcitonin gene-related peptide (CGRP) (26) and adenosine (28,29). On a cellular level, hypoxia-induced gene transcription factors (HIFs) are considered the master regulators of the hypoxic response (36). HIFs upregulate genes that promote survival during hypoxic conditions, such as glycolysis enzyme genes and vascular endothelial growth factor (VEGF). Some studies suggest that hypoxia, possibly via HIFs, causes disruption of the blood-brain barrier (BBB) (36) and oxidative stress via generation of reactive oxygen species (ROS) (37). If the aforementioned strategies fail to maintain adequate oxygen supply, the brain can produce energy anaerobically via glycolysis with the generation of lactate. During hypoxia, clinical studies with healthy individuals have shown increased lactate concentrations in the brain (38,39) as well as increased lactate release from the cerebral circulation (40). Studies have reported that lactate causes vasodilation mediated by extracellular adenosine (41) and via gap junction uncoupling under hypoxic conditions (42).
Hypoxia and migraine
An epidemiological study from the Peruvian Andes compared the prevalence of migraine in a mining population with permanent residence at high altitude (Cerro de Pasco, 4300 m, n = 1226), with a similar population at sea level (Lima, 330 m, n = 1031) (4). The migraine prevalence in the high-altitude population was higher (12% vs. 4%) than that of the sea-level population (4). Interestingly, the authors later focused on a group of 379 male miners in Cerro de Pasco and estimated the prevalence of migraine in this group to be 19% (5). The prevalence of migraine with aura (MA) was 12%, which is four times as high as the MA prevalence (3%) in males in the general population at sea level (43). Furthermore, MA was more prevalent than migraine without aura (MO) (12% vs. 7%) (43), which is the opposite of what one would expect at sea level (MA vs. MO, 5% vs. 8%) (5). Many factors have likely contributed to the high prevalence of migraine, as most of the high-altitude residents were miners, a profession exposed to dust, pollution and challenging working conditions. The study detected a significant correlation between working night shifts and extra hours, and frequency of headache. However, the female (non-miner) population also displayed a significantly higher prevalence of migraine in the high-altitude population, compared to the one at sea level (17.3% vs. 6.2%). Furthermore, the study found a higher prevalence of migraine in the higher age groups, which could be attributed to hypoxia-induced polycythemia (5). A different epidemiological study of an Andean high-altitude population (Cuzco, 3380 m), found a one-year prevalence of migraine of 5%, but a one-year prevalence of non-migraine headache of 29% (44). This population resided at a lower altitude than the first (3380 m vs. 4300 m). As such, the altitude, and consequently less severe level of hypoxia, may not have been sufficient to induce migraines. Furthermore, there was no control group in this study (44).
There have been several accounts of visual disturbances, particularly in people who suffer from migraines, induced at high-altitude conditions (45,46) and high-altitude altering the character of MA attacks (47). A prospective high-altitude study (45) followed 60 people (of whom 13 had a definite history of migraine) and an additional 19 people, at an altitude greater than 3000 m above sea level. The characteristics of the four worst headaches experienced by each person were recorded. Headaches experienced by people who suffered from migraines in daily life were more likely to be accompanied by phonophobia compared to headaches experienced by people who did not suffer from migraines in daily life (27% vs. 8%). A small study (n = 4) reported that migraineurs tended to report more severe HAH than non-migraineurs (48). A prospective altitude study of 506 mountaineers has shown that having a history of migraine is an independent risk factor for developing HAH (p < 0.01) (49). This association was also detected in a similar prospective study of 1326 mountaineers (50). Having a history of migraines appears to be an even greater risk factor for AMS, as a prospective study of 431 mountaineers found that suffering from regular migraines increased the risk of AMS 13 (4.3–39.2) times (51). However, the many similarities between the clinical manifestations of migraine, HAH and AMS make it difficult to determine whether these numbers reflect an increased susceptibility to secondary headaches among migraineurs, or genuine migraine attacks that are in fact provoked by the high-altitude environment.
These similarities have spawned investigations of the efficacy of sumatriptan as treatment of HAH and AMS (6–9). A small, uncontrolled study found that 100 mg of oral sumatriptan resulted in headache relief in five out of nine mountaineers within 30 minutes of intake. After 12–24 hours, the headache recurred in all except one person (6). A randomized, placebo-controlled, double-blind trial tested the efficacy of 100 mg oral sumatriptan in 29 mountaineers with moderate to severe headache. The study did not detect a difference between treatment groups (7). A small study (n = 33) (8) compared the efficacy of 100 mg oral sumatriptan against 600 mg oral ibuprofen in treating HAH. There was no difference between the groups, although ibuprofen tended to be superior to sumatriptan (8). A randomized, placebo-controlled, double-blind trial investigated the preventive effect of 50 mg oral sumatriptan taken within one hour of ascent (9). After 24 hours, AMS was more prevalent in the placebo group (n = 23 (45%)) than the sumatriptan group (n = 12 (24%); p = 0.02) (9). Furthermore, the rate of headache was higher among placebo users (placebo vs. sumatriptan group: 29 (57%) vs. 17 (33%); p = 0.02) (9). The aforementioned difficulty in differentiating between HAH, AMS and migraine attacks could perhaps explain some of the discrepancies between the results of these studies. Thus, although some studies point to an effect of sumatriptan in AMS and HAH, the evidence is sparse and no clear conclusion can be drawn.
In a clinical migraine provocation study, Schoonman et al. (10) investigated normobaric hypoxia as a possible trigger of migraine attacks. In this randomized, crossover, double-dummy study, 14 migraine patients were exposed to two different provocations (normobaric hypoxia and glyceryl trinitrate (GTN) infusion) and one sham provocation on three separate days. Normobaric hypoxia was induced by breathing oxygen-reduced air, equivalent to peripheral oxygen saturation (SaO2) values of 75%–80%, for up to five hours. Only six of the 14 patients experienced MO attacks after the hypoxia challenge, compared to two after the sham provocation. However, the migraine group may have been less sensitive to provocation, as only 21% experienced migraine attacks after GTN, which is a much smaller proportion than what other studies have reported (52,53). In a recent study (54), we exposed 15 MA patients to 180 minutes of normobaric hypoxia (mean capillary oxygen saturation 72%) in a randomized, sham-controlled, double-blind, crossover study design. Eight patients experienced migraine-like attacks during hypoxia compared to one patient after sham (p = 0.039) (54).
Broessner et al. (55) investigated whether normobaric hypoxia might trigger migraine-like headache attacks in healthy volunteers without any history of migraine or chronic tension-type headache. In total 77 volunteers were exposed to a fraction of inspired oxygen (FiO2) of 12.6% in a hypoxic chamber, equivalent to a simulated altitude of 4500 m. The SaO2, AMS severity score and headache characteristics were assessed at baseline, six hours and 12 hours. Eighty-one percent of the participants developed headache six hours after start of normobaric hypoxia. Twenty-seven percent (at six hours) and 43% (at 12 hours) of participants reported headaches with at least two migraine-like features. Interestingly, 8% and 15% of the participants experienced headaches, which fulfilled the International Classification of Headache Disorders, third edition beta (ICHD 3-beta) criteria for migraine-like headache after six and 12 hours, respectively. At time point six hours, SaO2 was a predictor for headache. These data suggest that normobaric hypoxia may induce headaches with migraine-like features in healthy volunteers without any history of migraine. However, given the young study population (mean age 26 years old), we cannot exclude that this study could have included people who may yet develop migraine. In addition, the study is limited by the fact that there was no control group and therefore the headache incidence might have been overestimated.
Few studies have investigated whether oxygen is an effective acute treatment in migraine. A recent Cochrane review (56) found that there was some evidence on effect of hyperbaric oxygen and insufficient data on efficacy of normobaric oxygen. A different review (57) concluded that the evidence regarding hyperbaric oxygen was unclear. Both reviews emphasized a need for further studies before clinical recommendations can be made (56,57).
Changes in barometric pressure have long been associated with headache and discussed as a possible migraine trigger (1,58). Studies suggest that quick reductions in barometric pressure (46,59) are more strongly associated with headache and migraine than slow reductions (60,61), which the body may adapt to. As such, air pressure changes may be a factor in itself or in consort with hypoxia in provoking headache and migraine.
Hypoxia and CH
Obstructive sleep apnea (OSA) is more common among CH patients (15,16) than the general population (62). One study reported that CH patients were 8.4 times more likely to experience OSA than a normal individual of equivalent age and body mass index (14). OSA is associated with hypoxemia. A sleep apnea and CH study observed that 57% of the CH attacks were preceded by an episode of desaturation (65%–85%) (16). One case report reported a 50% reduction in CH attack frequency during the first four years after beginning continuous positive airway pressure (CPAP) treatment, and hence treatment for the associated hypoxemia, in an episodic CH patient with OSA (63). This suggests that hypoxia may be a trigger for CH attacks. However, there are several potential confounders, as the correlation between OSA and CH could also be caused by poor quality of sleep or disruptions in sleep architecture.
Oxygen is highly effective as acute treatment for CH attacks. A large, randomized, double-blind, placebo-controlled trial found that high-flow (12 l/min) 100% oxygen therapy provided total pain relief or adequate relief after 15 minutes in 78% of participants compared to 20% with placebo (17). One hundred percent oxygen constitutes the first-line therapy for CH (64). A case report described an episodic CH patient whose attacks at altitude (1500 m) were non-responsive to her usual therapy (subcutaneous sumatriptan), but were aborted by oxygen (100% at 7 l/min.) after only five minutes (65).
Zhao et al. (66) exposed 15 CH patients in active period, 15 CH patients in remission and 15 control individuals to 12% O2 for 30 minutes, but only one CH patient experienced an attack during the provocation. The SaO2 was reduced gradually over 30 minutes from basal value to 82.4 ± 5.1% in the active-period CH patients, 79.6 ± 6.3% in the remission CH patients and 78.8 ± 4.3% in controls. In comparison, migraine patients were exposed to 75%–80% SaO2 for 4.5 hours (median) (10) and 72% SaO2 for 105 minutes (median) (54) before migraine attacks occurred. Two studies exposed episodic CH patients to transient hypoxia in active and remission period (67,68). In the first study, 20 CH patients (11 in active period, nine in remission) inhaled one to eight breaths (mean 3–4 breaths) of 100% nitrogen once, until the SaO2 decreased to approximately 80% (67). In the second study, 11 CH patients (six in active period, five in remission) inhaled 12% oxygen for five minutes (68). None of the participants developed CH attacks. Thus, hypoxia for a short duration did not induce CH attacks.
Possible hypoxic mechanisms in migraine and CH
NO
NO is an important molecule in migraine and CH pathophysiology (69). GTN, an NO-donor (70), triggers migraine attacks in migraineurs (52) and CH attacks in CH patients (71,72). It is possible that hypoxia-induced vasodilation and headache is mediated by NO. In a crossover magnetic resonance imaging (MRI) study, eight healthy volunteers were exposed to 80% hypoxia for 20 minutes and the CBF was measured noninvasively using a phase-contrast MRI technique in the basilar and internal carotid arteries (24). Hypoxia induced a 15% increase in CBF, which was reversed to baseline value by the competitive nonspecific NO synthase inhibitor L-NMMA (24). Deoxyhemoglobin appears to reduce nitrite to NO, which could be a key mechanism behind hypoxic vasodilation (73,74). During hypoxia, a net cerebral uptake of nitrite has been observed (75). Furthermore, the concentration of the O2 radical superoxide, a scavenger of NO, falls during hypoxia (76). As such, in conditions with less available oxygen, such as at high altitude, NO activity may be increased (69). Interestingly, a clinical double-blind crossover study with seven healthy males found that L-arginine (the substrate of NO) dietary supplementation after ascent to 4243 m increased headache scores (significantly at 12 hours) (77).
Other gases
Carbon dioxide (CO2) could be involved in migraine and headache pathophysiology. Intranasal CO2 application has been used clinically to treat migraines, and in vivo and in vitro rat studies suggest a possible effect on the trigeminal nerves (78,79). CO2 is involved in vasoregulation, and Liboni et al. (80) suggested that MA patients may have impaired CO2 regulation. Carbon monoxide (CO) may also play a role in headache and migraine, via cyclic guanosine monophosphate (cGMP) signaling, peripheral and central nociceptive processing, vasodilation and oxidative stress (81). Pollution studies (82–84) suggest that increased levels of nitrogen dioxide (NO2), CO and sulfur dioxide (SO2) show some association with the number of clinical headache and migraine visits. Future studies should investigate whether changes in these gases during hypoxic conditions could also contribute to triggering headache and migraine.
CGRP
CGRP is also an important molecule in migraine pathophysiology and can induce migraine attacks through cyclic adenosine monophosphate (cAMP) signaling. Whether CGRP can induce CH attacks is unknown. CH patients in active period have increased levels of CGRP in saliva (85) and in the external jugular vein ipsilateral to the pain ictally, compared to post-ictally and controls (86). Furthermore, Fanciullacci et al. (72) reported increased plasma CGRP levels during cluster attacks induced by GTN. Hypoxia potentiates the vasodilatory effect of αCGRP in isolated porcine intramyocardial arteries and increases the production of cAMP via αCGRP (26). Another in vitro study reported an upregulation of CGRP-receptor components during hypoxia (87). Studies of rat spinal cord slices and guinea pig hearts showed that lactic acid and low pH can stimulate and potentiate CGRP-release from sensory nerve terminals (88,89). As such, hypoxia could perhaps increase cerebral CGRP via increased lactate levels (38,39). In humans, a close linear correlation between the relative increase in plasma CGRP and lactate levels during exercise has been demonstrated (90). However, at rest there were no changes in CGRP levels after either 24 hours or five days at 4559 m hypobaric hypoxia in healthy volunteers (90).
Cortical spreading depression (CSD)
The CSD of Leao (91), a primary brain phenomenon, is generally accepted as the mechanism behind migraine aura (92–94). CSD is a self-propagating wave of neuronal depolarization (91,95) and is associated with a pronounced release of various neurotransmitters, vast shifts in ionic gradients and changes in blood flow (96). Animal studies suggest that hypoxia may be able to induce (97), extend (98,99) and lower the threshold for CSD (98). Functional imaging studies of humans (93,94) and rats (92) indicate that CSD is associated with an initial, short-lasting focal hyperemia followed by prolonged hypoperfusion. These CBF changes appear to be focal, localized to the outermost layers of the cortex and are not associated with general cerebral ischemia (100), and CSD does not appear to be associated with neuronal injury in healthy brain tissue (101).
Unlike MO, no validated human model to provoke and investigate migraine aura attacks exists. In our recent hypoxia provocation study (54), hypoxia induced migraine aura in three out of 15 MA patients and uncharacteristic visual disturbances categorized as possible aura in four more patients. These data suggest that hypoxia may provoke aura attacks in a subgroup of patients and that a hypoxia model may be used to investigate aura mechanisms under controlled conditions. Hypoxia can be used to elicit spreading depression-like depolarization in animal models (97). Mané and Müller (97) exposed isolated rat hippocampal slices suspended in oxygenated cerebrospinal fluid (CSF), to a severely hypoxic atmosphere (95% N2, 5% CO). Spreading depression-like phenomena were recorded using both direct current (DC)-potential and intrinsic optical signaling measurements. Takano et al. (98) found that CSD was associated with short periods of tissue hypoxia in watershed regions in live mice. They also detected an inverse correlation between the duration of CSD in rats and the availability of oxygen. This suggests that hypoxia increases the duration of CSD and that oxygen supply is the rate-limiting step for normalization of the extracellular concentration of K+ (98). Piilgaard and Lauritzen (102) observed that experimentally induced CSD caused an increase in the cerebral metabolic rate of oxygen of 71% for two to three minutes in rats. Despite a subsequent 238% rise in CBF, tissue oxygen tension decreased by 57%–100%. These findings may reflect a transient mismatch between excessive oxygen consumption and insufficient supply of oxygen during CSD due to neuronal uncoupling (98,102). Sonn and Mayevsky (99) detected similar results in anesthetized rats by using KCl to induce CSD under both normoxic and hypoxic (induced by inhalation of 12% oxygen for 17–20 minutes) conditions. Furthermore, they observed that the CSD wave was prolonged during hypoxia compared to normoxia. Thus, hypoxia is most likely not just an epiphenomenon during CSD, but can induce and intensify CSD. Furthermore, whether CSD provokes migraine attacks has not been definitively proven and is still a matter of debate.
Autonomic pathways and regulation
Oxygen inhalation is the standard acute therapy for cluster headache but the mechanisms underlying the antinociceptive effect of 100% oxygen in CH are poorly understood. One study (103) assessed the effect on the cranial parasympathetic system in a rat model of CH by stimulating the superior salivatory nucleus while monitoring neuronal firing in the trigeminocervical complex and blood flow to the lacrimal duct. Oxygen did not alter dural-evoked responses in the trigeminal nerve. However, 15-minute oxygen exposure inhibited superior salivatory nucleus-evoked responses by up to 33% in the trigeminocervical complex. Increases in concurrent blood flow in the lacrimal duct were also reduced by 15-minute oxygen administration. A later study by the same group observed similar results (104). This suggests that oxygen does not assert a direct effect on trigeminal afferents. Rather, it inhibits both evoked trigeminovascular activation and activation of the autonomic pathway by acting specifically on the parasympathetic/facial nerve projections to the cranial vasculature (103).
In the provocation study (66), the patients in active period showed a smaller decrease in SaO2 than the controls. A trend was reported when comparing patients during active and remission periods. The authors suggested that CH patients had an abnormality in the central autonomic regulation during the active period. However, the blood CO2 level was not measured and hyperventilation could not be ruled out. The CH patients in active period may have been more anxious about provoking an attack. Similarly, Kudrow and Kudrow (105) showed that GTN administration to 10 CH patients in active period, 10 in remission and five controls was followed by a decrease in arterial SaO2 in all participants. In the remission and control groups, the SaO2 then returned to baseline value. However, the active CH patients continued to have a lower SaO2, which culminated in CH attacks in all patients, while no attacks were reported in the remission or control groups. Kudrow and Kudrow (105) suggested that the association between CH attacks in CH patients during active period and an observed drop in SaO2 could be caused by a possible impaired autoregulation during the CH phase. However, Shen et al. (67,68) did not detect any differences in the ventilatory or heart rate response to hypoxia in episodic CH patients in active period and in remission compared to controls. It should be noted though that the hypoxic exposures in both studies were very short-lived (three to four breaths and five minutes, respectively).
Other possible mechanisms
In vivo and in vitro animal studies (36) suggest that hypoxia may lead to disruption of the BBB. This may be mediated by the HIFs (36). It has been proposed that migraine attacks are associated with disruptions in the BBB (106). One case-report study showed mild BBB disruption during a severe attack of a rare subtype of MA, familial hemiplegic migraine (107). To date, no firm evidence exists supporting the hypothesis on disruption of the BBB in common types of migraine.
Neurogenic inflammation has been suggested as a possible cause of activation and sensitization of peripheral nociceptors in migraine (108,109). It involves release of various pro-inflammatory mediators such as bradykinin, cytokines and oxidative stress (108,109). A transient increase in the pro-inflammatory cytokines tumor necrosis factor alpha (TNFα) and interleukin 6 (IL-6) in internal jugular blood has been reported during the initial two hours after the onset of MO attacks (110). The effect of oxygen on CH patients has been suggested to be caused by an inhibition of neurogenic inflammation (111). Schuh-Hofer et al. (111) assessed the effect of oxygen on neurogenic inflammation by examining plasma protein extravasation in the dura mater of rats. The study showed that the ratio of dural plasma protein extravasation was significantly lower under hyperoxic conditions compared to normoxic conditions. Furthermore, the association appeared to be dose dependent. In clinical studies, increased levels of the inflammatory response markers IL-6, C-reactive protein (CRP) and prostaglandins have been observed during hypobaric hypoxia (112,113).
Hypoxia is associated with oxidative stress in humans, especially during exercise (37,114,115). Many migraineurs report that exercise can trigger their attacks. However, Hougaard et al. were able to provoke attacks in only four (one MA and three MO) out of 12 MA patients using physical exercise (116). In addition, migraine has been associated with elevated levels of the oxidative stress biomarker 4-hydroxy-2-nonenal (HNE) interictally (109). ROS could be generated by hypoxia via inducible nitric oxide synthase (iNOS) (69) and HIFs (37).
It has recently been proposed that acid-sensing channels (ASICS) play a role in migraine pathophysiology (117). Increased concentrations of lactate in the brain (38,39) and cerebral circulation (40) have been detected during hypoxic exposure in healthy individuals.
Both acute (46,59) and slow (60,61) changes in air pressure, for instance, in relation to flying, has also been associated with secondary headache (1) and possibly migraines (58). As such, this could also contribute to triggering migraines at altitudes.
Conclusion
Hypoxia induces secondary headaches, which in phenotype and treatment response may mimic migraine, but whether the pathophysiological mechanisms are similar remains an open question. Thus, high-altitude epidemiological studies and sumatriptan-response studies in HAH and headache due to AMS do not clearly answer whether hypoxia simply triggers genuine migraine attacks in susceptible individuals at high altitude. CH’s relation to hypoxia is intriguing given the association with OSA and therapeutic response to oxygen inhalation, but provocation studies have not convincingly shown that hypoxia can induce CH attacks. Oxygen appears to be more effective in treating CH attacks than migraine attacks, suggesting different underlying mechanisms. Nevertheless, this review describes how hypoxia may be involved in liberation of NO and CGRP, changes in CO2, NO2 and CO, CSD, disruption of the BBB and neurogenic inflammation, which are mechanisms that have been proposed to occur during migraine and CH attacks. Human headache provocation models using hypoxia have yielded interesting results and are a novel approach to study in depth the mechanism underlying hypoxia and primary headaches. Future studies using human provocation models and neuroimaging along with experimental animal models may further elucidate the role of hypoxia in primary headaches.
Article highlights
Hypoxia causes secondary headaches such as high-altitude headache (HAH) and headache due to acute mountain sickness. Hypoxia-induced headaches mimic primary headaches such as migraine, which suggests a common link. Hypoxia is involved in liberation of nitric oxide (NO) and calcitonin gene-related peptide (CGRP), cortical spreading depression (CSD), disruption of the blood-brain barrier (BBB) and neurogenic inflammation, which are mechanisms underlying the pathophysiology of migraine and cluster headache. Human hypoxia models of headache provide a novel approach to study in depth the link between hypoxia and primary headaches.
Footnotes
Acknowledgment
The authors thank Anna Magnussen for the illustrations.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Capital Region of Denmark Foundation for Health Research (A4620), the Lundbeck Foundation (R155-2014-171), the Novo Nordic Foundation (NNF11OC1014333), the Augustinus Foundation (13-3794), Danish Council for Independent Research (DFF-4004-00169B), Simon Fougner Hartmanns Familiefond, and the European Union’s Seventh Framework programme (2007-2103) under grant agreement no. 602633.