
Abstract
Background
We studied lateral inhibition in the somatosensory cortex of migraineurs during and between attacks, and searched for correlations with thalamocortical activity and clinical features.
Participants and methods
Somatosensory evoked potentials (SSEP) were obtained by electrical stimulation of the right median (M) or ulnar (U) nerves at the wrist or by simultaneous stimulation of both nerves (MU) in 41 migraine without aura patients, 24 between (MO), 17 during attacks, and in 17 healthy volunteers (HVs). We determined the percentage of lateral inhibition of the N20–P25 component by using the formula [(100)–MU/(M + U)*100]. We also studied high-frequency oscillations (HFOs) reflecting thalamocortical activation.
Results
In migraine, both lateral inhibition (MO 27.9% vs HVs 40.2%; p = 0.009) and thalamocortical activity (MO 0.5 vs HVs 0.7; p = 0.02) were reduced between attacks, but not during. In MO patients, the percentage of lateral inhibition negatively correlated with days elapsed since the last migraine attack (r = −0.510, p = 0.01), monthly attack duration (r = −0.469, p = 0.02) and severity (r = −0.443, p = 0.03), but positively with thalamocortical activity (r = −0.463, p = 0.02).
Conclusions
We hypothesize that abnormal migraine cycle-dependent dynamics of connectivity between subcortical and cortical excitation/inhibition networks may contribute to clinical features of MO and recurrence of attacks.
Background
The exact pathophysiological mechanisms underlying the recurrence of migraine attacks are still not completely understood. Brainstem modulatory circuits and altered cortical excitability may play a pivotal role. In particular, clinical neurophysiology studies have shown that in most migraine patients between attacks cortical responsivity to various sensory stimuli, except olfactory ones, is enhanced compared to controls. This functional brain abnormality is not stable, but cyclically changes up to the tipping point of the attack when cortical responsivity normalizes (1).
There is as yet no single causal explanation for this dynamic cortical dysfunction and its precise role in recurrence of migraine attacks and its possible relation with clinical features of the disease are not known. An imbalance between inhibitory and excitatory cortical mechanisms, primary or secondary to reduced cortical pre-activation levels because of an insufficient thalamocortical drive, has been considered a possible culprit (1).
That the cortical hyper-responsivity in migraine could be due to abnormal thalamic control, and thus to so-called “thalamocortical dysrhythmia” (TCD), is suggested by studies of high-frequency oscillations (HFOs) in spontaneous electroencephalogram (EEG) and evoked potentials (2–4). This concept may reconcile the advocates of excessive excitation or deficient inhibition, since a deficient thalamocortical drive may result in a dysfunction of both inhibitory and excitatory cortical neurons (5). In line with the TCD hypothesis, we have recently shown that short-range lateral inhibition is impaired interictally in the visual cortex of migraine patients and that this impairment contributes to the hyper-responsivity of visual-evoked potentials (VEPs) found in migraine (6).
Among the various neurophysiological techniques, somatosensory-evoked potentials (SSEPs) were sensitive in disclosing abnormal cortical responsivity in migraine patients studied interictally (4). They also have the advantage of allowing reliable assessments of HFOs as indices of thalamocortical activation (7).
In humans, simultaneous stimulations of a pair of two adjacent peripheral nerves elicit smaller SSEPs than separate stimulations of either of the two nerves. Based on experimental models, mechanisms of lateral inhibition are thought to account for SSEP suppression after simultaneous dual input (8–10). To the best of our knowledge, the degree of lateral inhibition in the somatosensory cortex has not been studied in episodic migraine without aura (MO) nor its possible correlation with thalamocortical activities.
Hence, we designed the present study to determine if the degree of lateral inhibition in the somatosensory cortex differs between MO patients recorded between or during attacks and healthy volunteers (HVs). For this purpose, we compared the amplitude of the low-frequency (LF) SSEP components recorded over the parietal cortex after stimulating simultaneously the median (M) and ulnar (U) nerves with the arithmetic sum of the amplitudes of corresponding SSEP components elicited by stimulating each nerve separately. Thereafter, we analyzed the HFOs embedded in the SSEP obtained after median nerve stimulation in terms of an early burst thought to be generated by pre-synaptic thalamocortical afferents and a subsequent component reflecting post-synaptic cortical activation. We sought in particular whether there is a relation between the degree of lateral inhibition and thalamocortical activity, or between electrophysiological patterns and clinical features. We reasoned that, on the one hand, lateral inhibition and thalamocortical activity would be reduced between attacks, but normalized during an attack, and that, on the other hand, these possible electrophysiological abnormalities would depend on migraine clinical features.
Methods
Participants
Demographics and clinical features. Data expressed as mean ± SD.
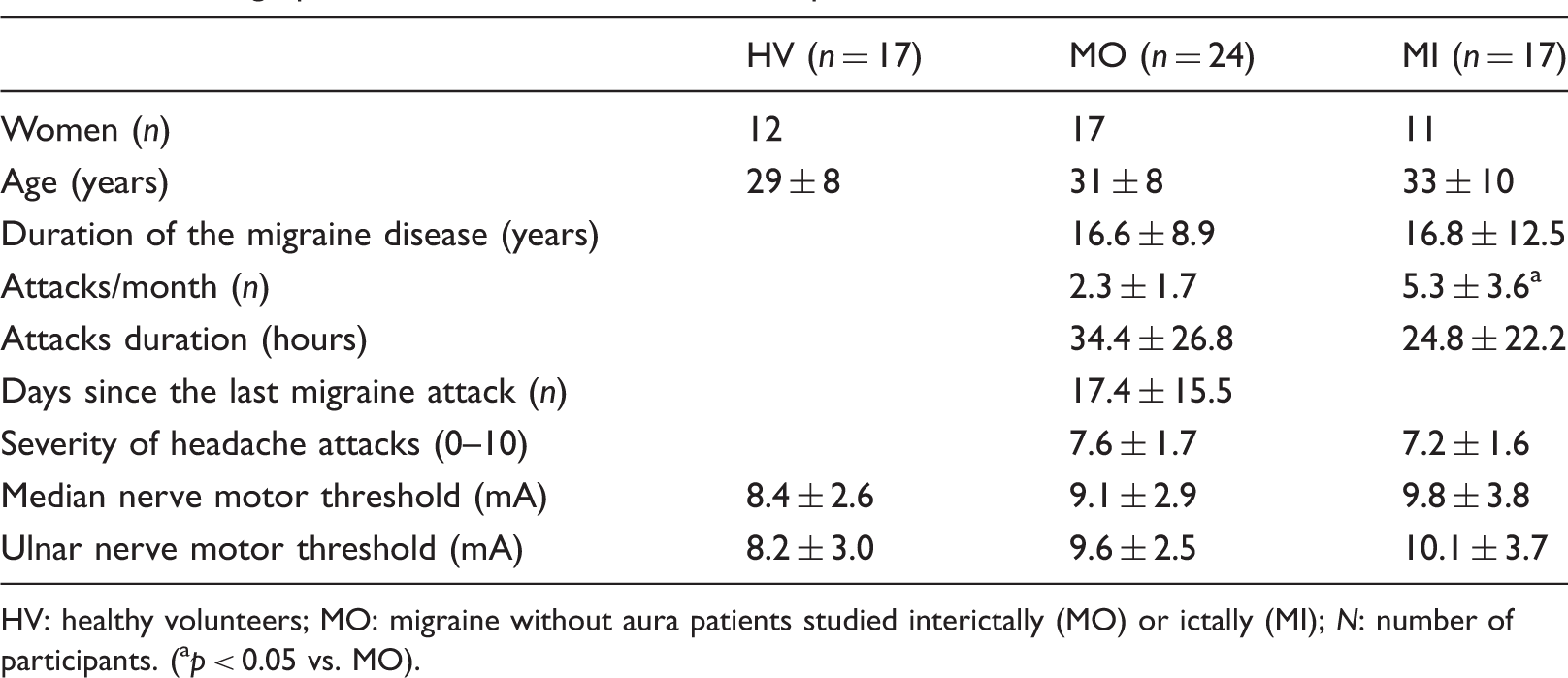
HV: healthy volunteers; MO: migraine without aura patients studied interictally (MO) or ictally (MI); N: number of participants. (ap < 0.05 vs. MO).
All patients fulfilled International Classification of Headache Disorders, third edition beta (ICHD-III beta) diagnostic criteria for episodic migraine without aura. Twenty-four patients (31 ± 8 y.o.; 17 women) were recorded during the interictal period (MO), i.e. at least three days before and after an attack, 17 patients (33 ± 10 y.o.; 11 women) during the ictal period (MI), i.e. from 12 hours before to 12 hours after an attack. The latter patients were not allowed to take acute medications before the end of recordings. We gathered information on various patient clinical characteristics by collecting two-month headache diaries at the time of either the screening visit and the day of the recording session: duration of the migraine disease (years), attacks frequency (n/month), attacks duration (hours), severity of headache attacks (0–10), and days elapsed from the last migraine attack (n) (Table 1).
For comparison, we recorded SSEPs in 17 HVs of comparable age and sex distribution (29 ± 8 y.o.; 12 women); they had no personal or familial history (first- and second-degree relatives) of migraine and no other detectable medical or psychiatric condition.
To avoid variability due to hormonal changes, women were recorded outside their premenstrual or menstrual periods.
Data acquisition
Three sets of SSEP recordings were performed in random order at ≥ 5-minute intervals by two investigators (MB and DDL): M nerve stimulation, U nerve stimulation and simultaneous stimulation of both nerves (MU). The stimuli were constant current square wave pulses (0.2 ms width, cathode positioned proximally), delivered at an intensity set at 1.2 times the motor threshold and a repetition rate of 2.2 Hz. The first active electrode was placed over Erb’s point ipsilateral to the stimulus referenced to the contralateral side; the second and third recording electrodes were positioned over the fifth cervical spinous process (Cv5) and over the contralateral parietal area (C3′, 2 cm posterior to C3 in the International 10–20 system), both referenced to Fz; the ground electrode was on the right arm. SSEP signals were amplified with a Digitimer™ D360 pre-amplifier (Digitimer Ltd, UK) (band-pass 0.05–2500 Hz, Gain 1000) and recorded with a CED™ power 1401 device (Cambridge Electronic Design Ltd, Cambridge, UK).
During the recordings carried out in the afternoon (between 2 p.m. and 6 p.m.), the participants sat relaxed in a comfortable chair in a well-lit room with eyes open. They were asked to fix attention on the stimulus-induced thumb movement. For each of the three stimulation protocols 300 consecutive sweeps of 50 ms, sampled at 5000 Hz, were collected. All recordings were analyzed off-line by one investigator (GC) blinded to the participants’ diagnoses using the Signal™ software package version 4.10 (CED Ltd). Artifacts were automatically rejected using the Signal™ artifact rejection tool if the signal amplitude exceeded 90% of analog-to-digital converter (ADC) range and controlled by visual inspection. The EP-signal was corrected off-line for DC-drifts, eye movements and blinks.
LF-SSEPs and lateral inhibition
For each of the three stimulation protocols, 300 artifact-free evoked responses were averaged in each individual. After digital filtering of the signal between 0 and 450 Hz, the various SSEP components (N9, N13, N20, P25 and N33) were identified according to their latencies. We measured peak-to-peak amplitudes of the peripheral N9 (recorded under the active Erb’s point electrode), the cervical N13 component (recorded under the active Cv5 electrode), and the cortical N20 (from the preceding positive peak), N20–P25 (peak to peak), and P25–N33 components (recorded under the active C3′ scalp electrode).
We calculated the percentage of lateral inhibition at the successive relays of the somatosensory pathway using the following formula: [100–(MU/(M + U)*100)], where MU is the amplitude of the SSEP component obtained after simultaneous stimulation of M and U nerves and M + U the arithmetic sum of the amplitudes obtained by stimulating separately these nerves.
SSEP HFOs
Using a method described elsewhere (4), digital zero-phase shift band-pass filtering between 450 and 750 Hz (Barlett-Hanning window, 51 filter coefficients) was applied off-line on SSEPs elicited by M nerve alone and by simultaneous stimulation of MU nerves in order to extract the HFOs embedded in the parietal N20 SSEP component. In the majority of recordings we were able to identify two separate bursts of HFOs: an early pre-synaptic burst within the latency interval of the ascending slope of the conventional N20 SSEP component and a late post-synaptic burst in the time interval of the descending slope of N20, sometimes extending into the ascending slope of the N33 peak. In general, the frequency of the oscillations was higher in the first than in the second burst and in between the pre- and post-synaptic bursts there was a clear frequency and amplitude decrease that allowed the separation of the two bursts. In recordings in which a clear distinction between the two components was not possible, we considered HFOs occurring before the N20 peak as pre-synaptic bursts and those after the N20 peak as post-synaptic bursts.
After eliminating the stimulus artifact, we measured the latency of the negative oscillatory maximum and the maximum peak-to-peak amplitude for each the two HFO bursts, and separately for M and MU stimulus conditions.
Statistical methods
We used the Statistical Package for the Social Sciences (SPSS) for Windows, version 21.0 for all analyses. The normal distribution of data for each group of individuals was assessed with the Kolmogorov-Smirnov test. A one-way analysis of variance (ANOVA) with group factor “subjects” (episodic migraineurs without aura studied ictally or interictally, and healthy participants) was used. Tukey test was used for post hoc analyses. Pearson’s correlation coefficient was used to search for correlations between lateral inhibition and the median nerve first HFO burst, as well as between SSEP amplitudes or lateral inhibition and clinical data such as severity of headache attacks (0–10), duration of migraine history (years), monthly attack frequency (n), attack duration (hours), and days elapsed since the last migraine attack (n). P values less than 0.05 were considered to indicate statistical significance.
Results
The demographics of recorded participants and the clinical characteristics of migraine patients are displayed in Table 1. This table also shows that there was no difference in motor thresholds after M or U nerve stimulation between the three groups of participants.
Assessable SSEP recordings were obtained from all patients and HVs participating in the study (Figure 1).
(a) Illustrative traces of low-frequency somatosensory evoked potentials (LF-SSEPs) showing N9, N13 and N20–P25 components after stimulating the median (M) or the median and ulnar nerves simultaneously (MU), and the arithmetic sum of responses obtained by stimulating each nerve separately (M + U) in a healthy volunteer (left traces), a migraine without aura patient recorded interictally (middle traces) and during an attack (right traces). (b) Illustrative traces of pre- and post- synaptic high-frequency oscillations obtained by digital band pass filtering (450–750 Hz) the median nerve SSEP responses in a healthy volunteer (HV), a migraine without aura patient recorded interictally (MO) or during an attack (MI).
LF-SSEPs and lateral inhibition
Latencies and amplitudes of the various SSEP components after median or ulnar stimulation (mean ± standard deviation; 300 averaged responses).

There are no significant differences between groups.
HVs: healthy volunteers; MO: migraine without aura patients between attacks; MI: migraine patients during an attack; SSEP: somatosensory evoked potentials.
Amplitudes of the various SSEP components (mean ± standard deviation) after simultaneous median and ulnar nerve stimulation (MU) or summed stimulation of these nerves (M + U) (300 averaged responses).

There are no significant differences between groups.
HVs: healthy volunteers; MO: migraine without aura patients between attacks; MI: migraine patients during an attack; SSEP: somatosensory evoked potentials.
ANOVA testing of the mean percentage of lateral inhibition [100–((MU/M + U)*100)] for brachial plexus N9, cervical spinal cord N13, and first cortical p-N20 components did not show a main effect for the factor group (N9 component: F (2,55) = 0.344, p = 0.710; N13 component: F (2,55) = 0.329, p = 0.720; p-N20 component: F (2,55) = 0.147, p = 0.862). By contrast, ANOVA for the mean percentage of lateral inhibition [100–((MU/M + U)*100)] of the cortical N20–P25 component disclosed a main effect for the factor group (F (2,55) = 4.227, p = 0.019). Post hoc analysis revealed that the lateral inhibition was significantly smaller in MO patients (27.9% ± 17.1; p = 0.024) than in HVs (40.2% ± 13.1), while in the MI group recorded during an attack it was similar (37.3% ± 10.0) to HVs (Figure 2).
Histogram showing the mean percentage of lateral inhibition (100–((MU/M + U)*100) for the N20–P25 component in healthy volunteers (HVs) and in migraineurs recorded interictally (MO) or ictally (MI). M: median; U: ulnar.
SSEP HFOs
Latencies and maximal amplitudes (mean ± standard deviation) of the first (pre-synaptic) and second (post-synaptic) HFOs embedded in median nerve SSEPs after stimulation of median (M) nerve alone and after simultaneous stimulation of the median and ulnar (MU) nerves.

M: Pre-synaptic HFO amplitude is significantly smaller in MO compared to HV (aANOVA p < 0.05).
There are no significant between group differences for post-synaptic HFOs.
MU: There are no significant between stimulus condition differences for pre- and post-synaptic HFOs in both MO and MI patients groups.
HFOs: high-frequency oscillations; HVs: healthy volunteers; MO: migraine without aura patients between attacks; MI: migraine patients during an attack; SSEP: somatosensory evoked potentials; ANOVA: analysis of variance.
After the simultaneous stimulation of the MU nerves, maximum peak-to-peak amplitudes and latencies of the negative oscillatory maximum of pre- and post- synaptic HFO were not different between groups (Table 4).
Correlation analyses
Pearson’s test disclosed several correlations between neurophysiological and clinical variables. Between attacks, Pearson’s test disclosed a negative correlation between N20–P25 SSEP amplitudes after separate stimulation of the M or U nerves (M: r = −0.442, p = 0.04; U: r = −0.597, p = 0.004) or their arithmetic sum (M + U: r = −0.529, p = 0.01) and days elapsed at the time of recordings since the last migraine attack (Figure 3). The percentage of lateral inhibition for the N20–P25 SSEP component [100–((MU/M + U)*100)] also correlated negatively with days from the last migraine attack (r = −0.510, p = 0.01, Figure 3), mean attack duration (r = −0.469, p = 0.02) and severity of migraine attacks (r = −0.443, p = 0.03, Figure 4), but it correlated positively with pre-synaptic M HFOs (r = 0.463, p = 0.02, Figure 5). Amplitude of pre-synaptic HFOs was negatively correlated with mean attack duration (r = −0.485, p = 0.01, Figure 5).
Correlation between the number of days elapsed since the last migraine attack and N20–P25 low-frequency somatosensory evoked potentials (LF-SSEP) amplitude (component N20–P25) after stimulating the median (M) or the ulnar (U) nerve separately, the arithmetic sum of median and ulnar nerve responses (M + U), and the mean percentage of lateral inhibition (100–((MU/M + U)*100) in migraine patients between attacks. Correlation between the mean percentage of lateral inhibition (100–((MU/M + U)*100) for the N20–P25 low-frequency somatosensory evoked potentials (LF-SSEP) component and severity of headache attacks (a), as measured by a visual analog scale (VAS), and mean attack duration (b) in migraine patients between attacks. M: median; U: ulnar. Correlation between amplitude maximum of pre-synaptic high-frequency somatosensory evoked potentials (SSEP) oscillations (HFOs), reflecting thalamocortical activity and the percentage of lateral inhibition (100–((MU/M + U)*100) for the N20–P25 low-frequency LF-SSEP component (left), or mean attack duration (right) in migraine patients between attacks. M: median; U: ulnar.


There was no significant correlation between neurophysiological and clinical data in MI patients recorded during an attack.
Discussion
Our study confirms previous findings showing that amplitude of pre-synaptic HFOs embedded in somatosensory evoked responses is reduced in episodic migraine between attacks, which is suggestive of diminished thalamocortical activity. By contrast, amplitudes of post-synaptic HFOs and of grand-average LF-SSEP components are within normal limits (4,11). An additional novel finding for SSEPs is that amplitude of the cortical N20–P25 component for all three stimulation paradigms is negatively correlated with time elapsed between the last migraine attack and the recordings, suggesting that amplitude of cortical SSEPs fluctuates with the migraine cycle, being minimal between attacks but increasing in proximity of or during an attack.
However, the most original aspect of our study is to show that lateral inhibition in the somatosensory cortex is reduced in MO patients between attacks, but normal during an attack. Moreover, we find that the degree of lateral inhibition is directly related with somatosensory thalamocortical activity, as indexed by amplitude of pre-synaptic HFOs, and inversely related to days elapsed since the last attack, severity of the migraine headache and attack duration. This suggests that reduced lateral inhibition in migraine is a consequence of poor thalamocortical drive, but also a marker of migraine severity.
Next to LF cortical SSEP representing the temporal marker of post-synaptic primary sensory cortex (S1) activation (12) and HFOs of SSEP reflecting thalamocortical fiber activity and primary cortical activation (7) it is possible to study lateral surrounding inhibition between neurons of the somatosensory cortex by stimulating adjacent nerves and the interaction between the afferent volleys produced by such simultaneous stimulation. It was shown indeed that in a subpopulation of neurones adequately activated by two different afferent inputs, the potentials recorded after simultaneous stimulation of two adjacent nerves are less ample than the arithmetic sum of the potentials generated by each stimulation separately (13,14). When converging inputs produce a response that is greater than the sum of the responses to each input in isolation, this is interpreted as due to lack of lateral inhibitory mechanisms (8). There is ample experimental evidence from electrophysiological single-unit recordings in animals that paired stimulations of the adjacent finger result in response suppression relative to single-digit stimulation, suggesting the presence of lateral inhibition across columns in layers two-thirds of area 3 b (9,10). Although SSEPs in supragranular layers are generated mainly by glutamate-mediated depolarization (15), they are under the inhibitory control of non-pyramidal GABAergic neurons (16). Among them, those with the largest lateral axonal field (>700 µm) are the putative neuronal mediators of short-range lateral inhibition between cortical columns (10). Given these data on the anatomo-physiological organization of the somatosensory cortex, our findings suggest that in migraine there is a cycle-dependent abnormal dynamic of the excitatory-inhibitory balance in response to peripheral somatosensory stimulation.
The various psychophysical and electrophysiological studies that have explored the central balance between inhibition and excitation in migraine are in line with this interpretation. In a pattern adaptation paradigm using visual illusions, a phenomenon attributed to a decrease in excitation from tonic pre-synaptic inputs, tilt after-effects, motion after-effects (17) and simultaneous tilt illusion (18) are prolonged in migraineurs as compared to HVs. This slow adaptation speed in migraine was attributed to a disequilibrium between excitatory and inhibitory mechanisms in the visual cortex (17–20). In a recent steady-state VEP study in which about 200 stimuli were delivered, supersaturation of VEP contrast gain was found in migraine between attacks and considered as supportive of excessive excitation driving increased network inhibition (21). When prolonged continuous stimulation is used, the pattern of response is different. In a refined steady-state VEP protocol using a windmill-dartboard (W-D) pattern of 600 stimuli, thought to explore preferentially short-range lateral inhibition, we have found a dual cortical response pattern: Lateral inhibition initially increases, in line with the VEP evidence mentioned above, while it decreases during the continuous stimulus repetition in MO but not in HVs (6). This observation is relevant to the present study that shows deficient lateral inhibitory mechanisms in the somatosensory cortex by analyzing grand-average SSEP between attacks after long-term stimulation, i.e. a condition quite similar to the final response pattern of W-D VEPs.
Another distinctive finding in our study is that the degree of lateral inhibition in the somatosensory cortex fluctuates over time in relation to the occurrence of a migraine attack: It is minimal at a long time interval after an attack while it is greater and within the range of normative values during an attack, which parallels the temporal dynamics of the LF-SSEP N20–P25 amplitude. The dynamic SSEP variations found here resemble those we have previously reported for VEPs after long-term stimulation, i.e. an interictal decrease as opposed to an ictal increase of lateral inhibition (6). In addition, a comparable correlation with the number of days elapsed between the last attack and the recordings was found in migraine for short-range lateral inhibition in the visual cortex (6), but also for visual metacontrast masking (22), functional magnetic resonance imaging (fMRI) blood oxygenation level-dependent (BOLD) response in the spinal trigeminal nuclei after intranasal nociceptive stimulation (23) or fractional anisotropy values in the thalamus (24). The latter is of particular interest for our finding of a strong positive correlation between thalamocortical activity and degree of lateral inhibition in MO patients: The lower the thalamocortical activity, the lesser the lateral inhibition. Globally, these results indicate that the overall subcortico/cortical drive changes with proximity of the last migraine attack.
A simultaneous dysfunction of thalamocortical activity and of cortical lateral inhibition is the hallmark of various functional brain disorders grouped under the name “thalamocortical dysrhythmia” (TCD) syndromes. The TCD theory proposes that a functional and anatomical disconnection of the thalamus from subcortical areas induces a change of rhythmic thalamocortical activity favoring cortical rhythms of lower frequency. This will reduce excitation of pyramidal cells at the beginning of the stimulus and of fast-spiking (FS) inhibitory interneurons during stimulus repetition (25). Converging evidence of abnormal thalamic/thalamocortical activity in migraine between attacks comes from the analysis of spontaneous (2) and evoked (3,4) rhythmic brain activity as well as from functional (26,27) and structural (24,28) neuroimaging studies. We have therefore hypothesized that migraine might belong to the thalamocortical dysrhythmias and that, in line with the TCD theory, low activity of the monoaminergic input to the thalamus and cortex might cause a functional disconnection of the thalamus leading to increased gamma band oscillations in the cortex and to reduced lateral inhibition (3).
Biochemical (29) and neuroimaging (30) studies indicate that in migraine monoaminergic transmission is altered. The brainstem aminergic nuclei modulate both the endogenous antinociceptive system and central processing of sensory stimuli by controlling preactivation levels and signal-to-noise ratio in cortical and thalamocortical neurons (31). As a matter of fact, the subcortical pain control systems seem to be hypoactive in migraine between attacks (32–34), while they may be hyperactive during the headache phase (35). An interictal dysfunction of the brainstem pain could thus perturb central sensory processing, and hence cortical metabolic homeostasis, and at the same time disinhibit the pain-signaling trigeminovascular system, hence paving the way for a migraine attack. Indirect evidence in favor of this hypothesis comes from our finding that the degree of lateral inhibition in the somatosensory cortex is inversely correlated with clinical features of migraine attacks reflecting its severity, such as intensity and duration of the headache (see Figure 4). The latter was also negatively related to thalamocortical activity, as assessed by amplitude of pre-synaptic HFOs, i.e. the greater the deficit of thalamocortical activation, the longer-lasting the headache attacks. That a worse clinical manifestation of migraine may be associated with low thalamocortical drive activity is also supported by the previous observation that patients who experience an increase in the number of migraine attacks during the six months following the recording session thalamocortical drives are reduced with respect to those who showed an improvement of migraine state (36).
Finally, we acknowledge as a limitation of the present study that not all the patients in the two groups (MO/MI) served as their own baseline reference, making impossible an immediate generalizability of our findings. Following the same patients longitudinally would have provided more robust statistical power. However, this point should not be considered detrimental because the two groups of patients (MO/MI) were recorded regardless during clearly distinctive periods during the so-called migraine cycle.
Conclusions
In conclusion, we have shown that lateral inhibitory mechanisms are impaired between attacks in the somatosensory cortex of migraine patients. We found that this impairment decreases in proximity to an attack, that it is associated with decreased thalamocortical activity and that it is directly correlated with the intensity and duration of usual migraine attacks. We propose that these data suggest that the aminergic brainstem-modulating systems are hypofunctioning between attacks and thus, either directly or indirectly via the thalamocortical loops, perturb both cortical information processing and endogenous pain control predisposing the migraineur to a migraine attack.
Future work should attempt to clarify the role of the somatosensory lateral inhibition mechanisms with regard to habituation/sensitization phenomena in episodic and chronic migraine.
Clinical implications
Mechanisms of lateral inhibitory are impaired between attacks in the somatosensory cortex of migraine patients. This impairment decreases in proximity to an attack, is associated with low thalamocortical activity and is directly correlated with the intensity and duration of usual migraine attacks.
Footnotes
Funding
This work was supported by the Italian Ministry of Health and Fondazione Roma.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.