
Abstract
Aim
The aim of this article is to evaluate the safety and efficacy of perimenstrual telcagepant, a CGRP receptor antagonist, for headache prophylaxis.
Methods
We conducted a randomized, double-blind, placebo-controlled, six-month trial in women with migraine for ≥3 months who experienced perimenstrual headaches. Women were randomized to telcagepant 140 mg or placebo (2:1 ratio) for seven consecutive days perimenstrually. Safety was assessed by adverse events and laboratory tests. The primary efficacy endpoint was mean monthly headache days in the subset of women reporting perimenstrual migraine (−2 days to +3 days of menses onset) and ≥5 moderate or severe migraines per month prior to entering the trial.
Results
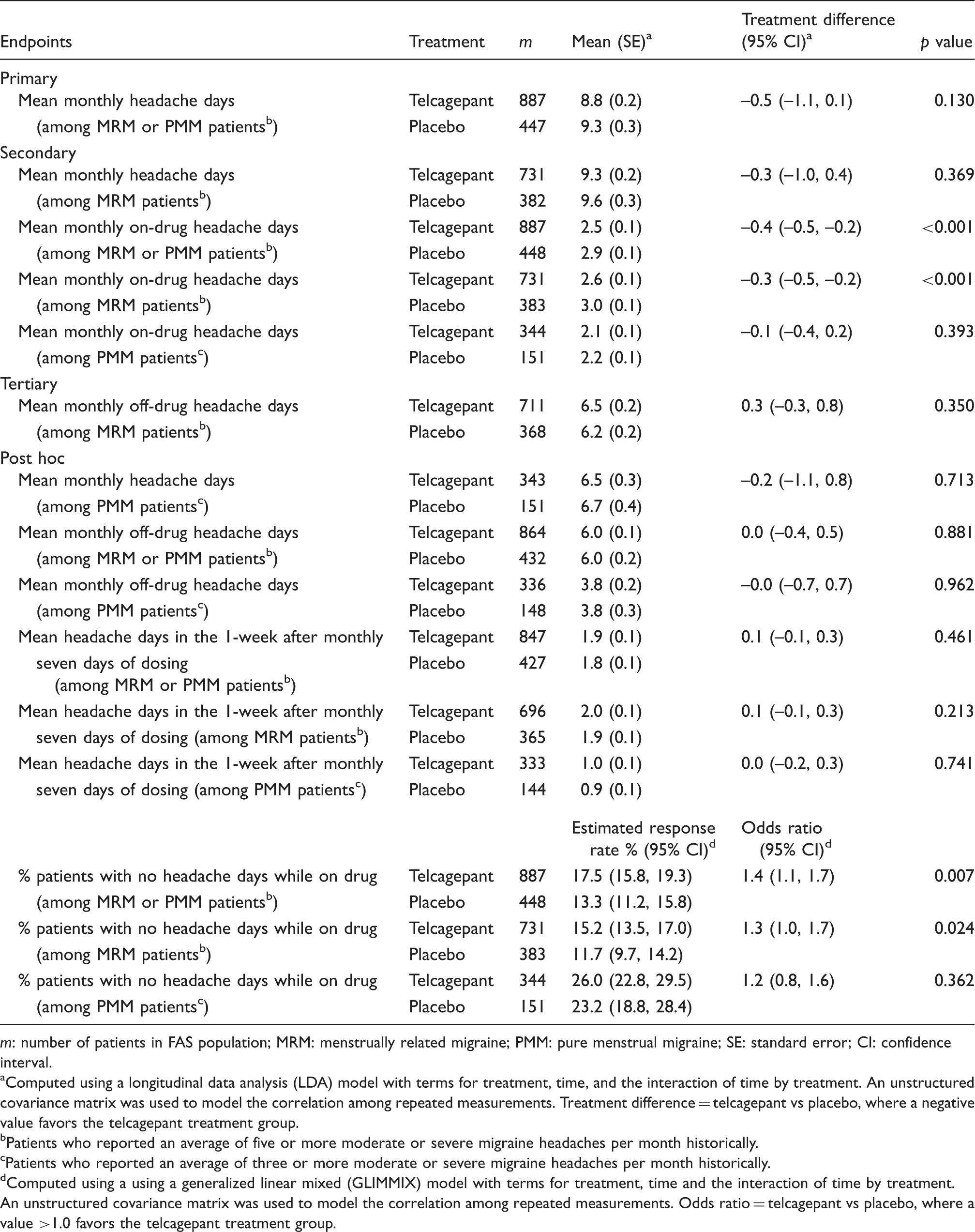
Telcagepant was generally well tolerated: 66/2660 (2.5%) on telcagepant and 36/1326 (2.7%) on placebo discontinued because of a clinical adverse event. The percentages of patients with clinical adverse events, laboratory adverse events, or discontinuation because of a laboratory adverse event were also similar between treatments. Alanine aminotransferase elevations ≥3× normal occurred in 0.6% of women on telcagepant and 0.4% on placebo. Three women on telcagepant vs none on placebo had alanine aminotransferase elevations ≥8× normal. In the efficacy subset there was no significant effect of telcagepant (n = 887) vs placebo (n = 447) in mean monthly headache days (treatment difference −0.5 day (95% CI: −1.1, 0.1)). However, telcagepant was associated with a reduction in on-drug headache days (treatment difference −0.4 day (95% CI: –0.5, –0.2), nominal p < 0.001).
Conclusions
Telcagepant 140 mg taken perimenstrually for seven days was generally well tolerated, but was associated with transaminase elevations. Telcagepant did not reduce monthly headache frequency, but did reduce perimenstrual headaches.
Introduction
Antagonism of calcitonin gene-related peptide (CGRP) receptors represents a novel approach to the treatment of migraine (1,2). Clinical trials have demonstrated that CGRP receptor antagonists have comparable acute efficacy to triptans (3–5). In Phase 3 trials, telcagepant, the most extensively studied CGRP receptor antagonist, was effective at doses of 140 mg and 280 mg, with the weight of evidence suggesting greater efficacy at the higher dose (5–9). A recent prophylaxis study also suggested that twice-daily administration of telcagepant 140 mg or 280 mg was effective for preventing migraines over one month (10). However, that study was terminated early because of safety concerns related to increased aminotransferase levels in some patients, and few patients received the planned three months of treatment.
The increased aminotransferase levels and potential for hepatotoxicity observed in the prophylaxis study precluded further development of telcagepant for chronic daily use. However, review of the telcagepant safety database suggested no increased aminotransferase elevation beyond the expected background rate of other therapies when use was for less than 14 consecutive days. Provided that these data were confirmed in a further study with a sufficiently large group of patients, telcagepant could be a viable treatment option in acute settings with a restriction on the maximum number of consecutive days of dosing or for short-term prevention.
To address this, a trial in women with perimenstrual migraine was designed that incorporated two elements. Firstly, we sought to obtain safety data on telcagepant 140 mg when used at an anticipated recommended maximum frequency of seven daily doses per month. A sufficiently large sample size was employed to assess the risk of aminotransferase elevation. Secondly, we sought to test the hypothesis that perimenstrual use of telcagepant 140 mg for seven consecutive days per month might be effective in preventing headaches. A low dose of telcagepant was chosen to attempt to minimize the risk of aminotransferase elevations.
Methods
Full details of the study methods and statistical analysis are provided in the study protocol, which is available as Supplementary material.
Patients
Women ≥18 years of age with a history of migraine with or without aura according to the International Classification of Headache Disorders, second edition (ICHD-II) criteria (11) for ≥3 months and ≥2 migraine attacks per month in the two months prior to screening were eligible. Patients were also required to report headache during the perimenstrual period (–2 days to + 3 days of menses onset) in ≥2 of the last three cycles, as well as regular menstrual cycles for the three cycles prior to study entry (an individual patient’s periods had to be approximately the same number of days each month and, in addition, the interval had to be between 22 and 32 days).
Patients were excluded if they had taken medication for an acute headache on ≥15 days per month in any of the three months prior to screening or were taking migraine prophylactic medication where the prescribed daily dose had changed during the four weeks prior to screening. Patients with aspartate aminotransferase (AST), alanine aminotransferase (ALT), or bilirubin levels that were >1.5× the upper limit of normal (ULN) or serum creatinine that was >2× ULN at a laboratory screening visit were excluded. Additional entry criteria and medication restrictions are described in the study protocol.
Patients were recruited at neurology/headache, general practitioner/family medicine, and gynecology/women’s health clinics. Patients were recruited mainly through the site’s databases of migraine patients, supplemented by advertisements to the general public and patient recruitment vendors. Patients received financial reimbursement for travel and inconvenience. All payments were disclosed and reviewed/approved by each site’s institutional review board.
Regulatory and ethical matters
The study was conducted in accordance with the principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agency. Each patient provided written informed consent. The study was registered at ClinicalTrials.gov (NCT01125774).
Study design
This randomized, double-blind, placebo-controlled, parallel-group study (Merck protocol 065) was performed at 229 investigative sites in the United States (US), Europe, Mexico, Australia and New Zealand from June 2010 to April 2011. Study eligibility was assessed at a screening visit, following which eligible patients were allocated in a 2:1 ratio to telcagepant 140 mg or placebo at bedtime, for seven consecutive days each month, beginning at the onset of menses, for up to six months. If patients could reliably predict the onset of menses they could begin dosing up to three days prior to menses onset.
Patients were allocated in a double-blind fashion using a computer-generated randomized allocation schedule prepared by a blinded statistician at Merck using a block size of six. Numbered containers were used to implement allocation. Personnel at each study site used a central interactive voice response system to determine which container should be given to which patient. All study personnel, including investigators, study site personnel, patients, and Merck staff remained blinded to treatment allocation throughout the study. Unblinding took place after data collection was complete.
Procedure
At the screening visit, patients completed a migraine history questionnaire to help determine eligibility (see above) and to categorize patients for the efficacy analyses (see below). During the trial, patients completed a daily paper diary on which they recorded presence of headache with duration ≥30 minutes (used to define a “headache day” for the efficacy analyses) and any adverse experiences. Other headache/migraine-related data were recorded in the diary (e.g. presence of aura and associated symptoms, maximum pain intensity, use of acute headache medication) but were not used in the present analysis. Patients returned to the clinic each month. At each clinic visit, patient diaries including adverse experiences were reviewed and routine laboratory tests and vital sign assessments were performed.
An external and independent safety monitoring board provided safety oversight and reviewed interim safety analyses at prespecified intervals during the study. Cases of AST or ALT elevations ≥3× ULN were adjudicated by an external blinded Aminotransferase Elevation Events Adjudication Committee. Confirmed elevations ≥3× ULN were defined as ALT or AST ≥3× ULN on initial and repeat testing within 72 hours, but if no repeat values were available the initial elevated value was considered a confirmed elevation. The adjudication committee determined relatedness of the elevation to the study drug, and whether there was a confounding factor(s) that could have contributed to, or accounted for, the elevation in aminotransferase(s).
Safety analyses
Tolerability and safety were summarized and assessed by statistical and clinical review of safety parameters, including adverse events, laboratory values, electrocardiograms (ECGs), and vital signs. The primary approach for safety analyses was the all-patients-as-treated approach. All patients who received at least one dose of study therapy were included in the treatment group according to the treatment they received. Although assessment of safety was a primary objective of the trial, the trial protocol did not designate any specific safety endpoints as “primary” (see Supplemental material). The trial registration site clinicaltrials.gov does not accept nonspecific objectives as outcome measures and therefore the following four safety outcomes were selected to represent the objective of safety assessment for trial registration: 1) percentage of patients with clinical adverse events, 2) percentage with discontinuations due to clinical adverse events, 3) percentage with laboratory adverse events, 4) percentage with discontinuations due to laboratory adverse events. These outcomes are listed as primary endpoints on clinicaltrials.gov but were not pre-specified endpoints in the trial protocol.
Efficacy analyses
It was planned that the efficacy analyses would be based on a minority subset of the total randomized population. Patients were classified as menstrually related migraine (MRM) or pure menstrual migraine (PMM) based on their responses to the following two questions on the migraine history questionnaire: 1) “For your past three menstrual cycles, did at least one migraine occur from the two days before menses onset through the first three days of menses in at least two out of three menstrual cycles?” and 2) “Over the last three cycles, did you get any migraines at other times?” MRM patients were defined as those who answered “yes” to both questions. PMM patients were defined as those who answered “yes” to question 1 and “no” to question 2. The time window of –2 to +3 days of menstruation was based on ICHD-II criteria for menstrual migraine (11). The primary efficacy endpoint was mean monthly headache days (headache duration ≥30 minutes) during the entire study period among the subset of MRM and PMM patients who reported that they experienced on average ≥5 moderate or severe migraine headaches per month in the two months prior to entering the study; that is, the intention was to evaluate the full effect of telcagepant on all headaches during the entire month. The cut-off of five was to guard against a ceiling effect in the reduction of migraine frequency and was in line with the population studied in a topiramate migraine prophylaxis study that had a mean of 5.5 migraine headaches per month at baseline (12). This primary endpoint was originally a secondary endpoint but was switched to the primary endpoint after the trial was initiated and before unblinding. The change was based on data suggesting the possibility of effects prolonged beyond acute exposure (5,10,13). The original primary endpoint was based on on-drug data and was moved to a secondary objective (see below). The primary efficacy analyses were based on all patients who were randomized, took ≥1 dose of study medication, and had ≥1 post-randomization efficacy measurement. Patients were included in the treatment group to which they were randomized. A longitudinal data analysis method (14) was used to analyze the data. The model included each post-baseline measurement during the individual monthly periods (Month 1 to Month 6) as response variables. The repeated-measures model included terms for treatment, time, and the interaction of time by treatment. An unstructured covariance matrix was used to model the correlation among repeated measurements. The treatment difference in terms of mean monthly headache days during the entire six-month treatment period as well as in each month was estimated and tested from this model.
Additional analyses were conducted to more fully assess the hypothesized migraine prophylaxis effect. The secondary efficacy endpoints were: 1) mean monthly headache days during the entire study period among MRM patients who reported that they experienced on average ≥5 moderate or severe migraine headaches per month in the two months prior to entering the study; 2) mean monthly on-drug headache days among MRM and PMM patients who reported that they experienced on average ≥5 moderate or severe migraine headaches per month in the two months prior to entering the study (the original primary endpoint when the trial was initiated); 3) mean monthly on-drug headache days among MRM patients who reported that they experienced on average ≥5 moderate or severe migraine headaches per month in the two months prior to entering the study; 4) mean monthly on-drug headache days among PMM patients who had reported ≥3 moderate or severe migraine headaches per month in the two months prior to entering the study. The tertiary efficacy endpoint was mean monthly off-drug headache days among MRM patients who had on average ≥5 moderate or severe migraine headaches per month at baseline. Analysis of the secondary and tertiary endpoints used the same methods as for the primary analyses. The prespecified strategy to adjust for multiplicity stipulated that the primary hypothesis had to be significant at the α = 0.05 level (two-tailed test) in order for the secondary hypotheses to be formally tested. Nominal p values were calculated for all comparisons.
A number of post-hoc analyses were performed including mean monthly off-drug headache days among PMM patients, the percentage of patients with no headaches per month while on the drug, and the mean headache days in the week immediately following the seven-day perimenstrual course of telcagepant each month (to explore if there was evidence of rebound headache after stopping telcagepant).
The data were analyzed using the SAS version 9.1 software package (SAS Corporation, Raleigh, NC, USA).
Power
The sample size was determined by safety requirements for a sufficient duration/number of patients exposed in agreement with the US Food and Drug Administration (FDA) rather than statistical power considerations. A 2:1 ratio was planned with ∼3000 and ∼1500 patients randomized to telcagepant and placebo, respectively. The power to reject the primary efficacy null hypothesis was >99%, assuming the true treatment difference in mean monthly headache days is one with a standard deviation (SD) of 4.6 and at least 40% of all randomized patients would contribute to the efficacy analysis (i.e. approximately 40% of randomized patients would have on average five or more moderate or severe migraine headaches per month at baseline).
Interim analysis
An independent safety monitoring board reviewed a formal unblinded interim analysis of the available safety data approximately one month after 1700 patients had been randomized. The independent safety monitoring board recommended that the study should continue.
Results
Patient accounting
The trial profile is shown in Figure 1. A pattern of patients enrolling at multiple sites was detected when one patient returned study medication dispensed at a different site. The clinical monitoring team then checked for patients with duplicate initials/dates of birth across the sites. Twenty-eight of the 4548 randomized patients (0.6%) were found to have enrolled multiple times at different sites (24 enrolled two times, three enrolled three times, and one enrolled four times). Of the 4520 women randomized excluding duplicates (telcagepant = 3018, placebo = 1502), 88% (3960) took treatment, and 72% of those taking treatment in each group completed (telcagepant = 1893/2638, placebo = 948/1322). The proportions of women in each discontinuation category were similar across treatment groups.
Study flowchart excluding 28 duplicate patients who enrolled at multiple sites. These patients were excluded from efficacy analyses but were included in the safety analyses as shown.
Patient characteristics
Baseline characteristics of treated patients: number (%) of patients except where stated.

US: United States; NSAID: nonsteroidal anti-inflammatory drug; h: hours.
Includes duplicate patients enrolled at multiple sites (see study flowchart). One duplicate patient who was originally assigned to placebo but switched to telcagepant after one month is counted in the placebo group for this table.
Perimenstrual period = from the two days before menses onset through first three days of menses.
Data available for 1515 patients in the telcagepant group and 782 patients in the placebo group.
Where data are missing, values do not add up to the number of treated patients and percentage values do not add up to 100%.
General tolerability
Summary of adverse events (AEs): number (%) of patients.

Listed as a primary safety outcome for the clinicaltrials.gov trial registration, but not pre-specified as such in the trial protocol.
Determined by the investigator to be related to the drug.
Patients who took both study treatments are counted in the telcagepant group, and only adverse events attributed to telcagepant are included.
Hepatic effects
Among treated patients who had at least one post-baseline ALT or AST test (N = 2660 for telcagepant, N = 1326 for placebo), the observed incidence of ALT or AST elevation ≥3× ULN was 17 (0.6%) in the telcagepant group and five (0.4%) in the placebo group. Confirmed elevations in ALT or AST ≥3× ULN were observed in 13 patients for ALT (10 (0.4%) for telcagepant and three (0.2%) for placebo) and seven patients for AST (four (0.2%) for telcagepant and three (0.2%) for placebo). None of these patients experienced a concomitant elevation in bilirubin >2× ULN. In three patients, all in the telcagepant group, ALT elevations ≥8× ULN were reported and were considered to be a serious laboratory adverse event; the elevation resulted in treatment discontinuation in one patient. In all three cases the elevations resolved or were resolving at the last study follow-up. The Aminotransferase Elevation Events Adjudication Committee determined that there were possible confounding factors in all three cases (unconfirmed positive hepatitis C test, positive hepatitis E serology, and acute cholecystitis) although none were sufficiently definitive to exclude the possibility of a treatment-related effect. The adjudication committee’s determination of the likelihood that the ALT elevations were related to study medication in these three patients, after taking into account the possible confounding factors, were “probably” (≥50% probability) in the patient with unconfirmed hepatitis C, “possibly” (25–50% probability) in the patient with hepatitis E, and “unlikely” (<25% probability) in the patient with cholecystitis. In the 17 telcagepant patients with ALT elevations ≥3× ULN, subgroup analyses of various factors associated with drug-induced liver injury (age, concomitant use of oral contraceptives, concomitant use of acetaminophen, and liver injury risk factor) failed to identify additional risk factors.
Based on an analysis of all treated patients, mean ALT levels were higher in the telcagepant group vs the placebo group by Month 1 (mean (SD) change from baseline: 0.8 (7.8) Iu/l for telcagepant and 0.3 (7.1) Iu/l for placebo) and continued through Month 6 (mean (SD) change from baseline: 1.9 (10.9) Iu/l for telcagepant and 1.2 (10.0) Iu/l for placebo). Mean AST levels were similar in the telcagepant group vs the placebo group at Month 1 (mean (SD) change from baseline: 0.2 (6.1) Iu/l for telcagepant and 0.0 (6.0) Iu/l for placebo) and through Month 6 (mean (SD) change from baseline: 0.6 (7.8) Iu/l for telcagepant and 0.5 (7.2) Iu/l for placebo).
Efficacy
A total of 887 patients for telcagepant and 447 patients for placebo, approximately a third of the overall sample, met the minimum migraine headache frequency criterion for inclusion in the primary efficacy dataset and were included in the analysis of the primary efficacy endpoint (Figure 1). Efficacy findings are summarized in Table 3. No significant difference between treatments was seen on the primary endpoint of mean monthly headache days. The lack of significance on the primary endpoint precluded the results of comparisons corresponding to the secondary hypotheses from having the possibility of formal statistical significance. However, based on nominal p values, there was a benefit of telcagepant over placebo when looking at monthly on-drug headache days. This benefit appeared to be driven by the MRM group rather than the PMM group (Table 3), and amounted to a difference of approximately half a day. The apparent efficacy in the MRM group was consistent over each month in the study (Figure 2). There was a similar pattern of findings in the post-hoc analyses including the percentage of patients with no headaches per month while on the drug. The post-hoc analysis of mean headache days in the week immediately after the seven-day perimenstrual course of telcagepant each month did not suggest evidence of rebound headache after stopping telcagepant (Table 3).
Mean (+ standard error) monthly on-drug headache days among menstrually related migraine (MRM) patients who historically reported five or more moderate or severe migraine headaches per month (secondary endpoint). Summary of efficacy of treatment on headache occurrence over the entire treatment period (average of the monthly values at each of Months 1–6). m: number of patients in FAS population; MRM: menstrually related migraine; PMM: pure menstrual migraine; SE: standard error; CI: confidence interval. Computed using a longitudinal data analysis (LDA) model with terms for treatment, time, and the interaction of time by treatment. An unstructured covariance matrix was used to model the correlation among repeated measurements. Treatment difference = telcagepant vs placebo, where a negative value favors the telcagepant treatment group. Patients who reported an average of five or more moderate or severe migraine headaches per month historically. Patients who reported an average of three or more moderate or severe migraine headaches per month historically. Computed using a using a generalized linear mixed (GLIMMIX) model with terms for treatment, time and the interaction of time by treatment. An unstructured covariance matrix was used to model the correlation among repeated measurements. Odds ratio = telcagepant vs placebo, where a value >1.0 favors the telcagepant treatment group.
Discussion
Our trial provided safety data in a sample of 2660 women for telcagepant and 1326 for placebo and is the largest safety trial performed to date for a CGRP receptor antagonist. In the majority of women, perimenstrual telcagepant for seven days per month was generally well tolerated over six months. These findings are in agreement with other Phase 3 acute efficacy studies in which telcagepant was used to treat a single migraine attack or multiple migraines intermittently over up to 18 months (5–9). A previous prophylaxis trial involving twice-daily dosing with 140 mg or 280 mg of telcagepant was terminated early because of ALT elevations in 13 patients receiving telcagepant but none on placebo (10). In the present trial of once-daily dosing with telcagepant 140 mg for seven consecutive days per month, overall rates of ALT and AST elevations were low but three patients in the telcagepant group vs none in the placebo group had ≥eight-fold normal ALT elevations without a definitive alternative explanation. This is consistent with the overall findings across the telcagepant development program that suggest that telcagepant is associated with increased risk for hepatotoxicity that appears to be related to the dose, frequency and duration of treatment, but that may be present even at low doses and limited treatment duration. The question of whether the hepatoxicity risk is a mechanism-based (CGRP class) effect or molecule specific is unresolved at present. Development of another CGRP receptor antagonist, MK-3207 (15), was terminated following the observation of liver test abnormalities in some patients but the pattern of findings was different from the findings for telcagepant; MK-3207 elevations generally occurred weeks after participants had stopped taking treatment and in some cases required immunosuppressive therapy whereas telcagepant elevations occurred during treatment and resolved on discontinuing treatment (10).
Our trial also evaluated the hypothesis that use of a CGRP receptor antagonist in the perimenstrual period might be effective in preventing headaches in women with perimenstrual migraine. The results showed that perimenstrual telcagepant 140 mg given for seven days each month was ineffective on the primary efficacy endpoint of reduction in mean monthly headache days in the subset of treated MRM and PMM patients (887 for telcagepant and 447 for placebo) with on average ≥5 moderate or severe migraine headaches per month. In secondary analyses, however, there did appear to be a modest, nominally statistically significant benefit of telcagepant of approximately 0.4 headache days per week compared to placebo when considering only on-drug headache days. This could be a chance finding given the number of comparisons performed. By extrapolation, and assuming that the menstrual relatedness of a headache has no impact on efficacy, this would suggest a reduction of approximately one to two headache days per month with daily dosing, which is consistent with efficacy findings from previous migraine prophylaxis studies (12,16).
The literature on menstrual migraine suggests that menstrual migraines are rarely associated with aura (11,17–19). It is therefore surprising that 40% of the patients in our sample reported that, by history, their migraines were usually preceded by aura. It is possible that some patients’ responses regarding aura included premonitory symptoms rather than being strictly migraine aura as there was no confirmation of what symptoms patients considered to constitute aura. Further, menstrual migraines can be without aura in women who have migraines with aura at other times of the cycle (20,21).
Our data are consistent with the prophylactic migraine effect of CGRP receptor antagonism seen in a previous trial with twice-daily dosing of telcagepant 140 mg and 280 mg in a general migraine population (10). The data suggest that CGRP receptor antagonists have an effect on headaches only while the drug is present. Thus the previous initial data suggesting the possibility of effects prolonged beyond acute exposure (5,10,13), which prompted the change of the primary efficacy endpoint after the study was initiated, may not represent true drug effects. As noted above, because of safety concerns about hepatotoxicity at higher doses, the dose of telcagepant evaluated here (140 mg daily for seven consecutive days) was lower than the doses evaluated in the previous migraine prophylaxis trial (140 mg or 280 mg twice daily), and it is possible that higher daily dose regimens might have shown greater efficacy.
In conclusion, our data suggest that CGRP receptor antagonists have some beneficial activity for preventing headaches during the time that patients are on the drug. However, the maximum magnitude of this effect with a CGRP receptor antagonist that could be used at an optimal dose is unknown. The development of telcagepant as an acute or prophylactic treatment for migraine has been discontinued by Merck.
Footnotes
Trial registration
ClinicalTrials.gov NCT01125774
Clinical implications
Calcitonin gene-related peptide (CGRP) receptor antagonists may have some beneficial activity for preventing headaches, but likely only during the time that patients are on the drug.
It is currently unclear whether the hepatoxicity risk associated with telcagepant is a CGRP class effect or is molecule specific.
Funding
This work was supported by Merck & Co. Inc, Kenilworth, NJ.
Conflicts of interest
TWH, APH, JG, CA, RG, CL and DM are current or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co. Inc, Kenilworth, NJ, and own or owned stock/stock options in Merck.
EAM served as a member of Merck’s International Migraine Advisory Board. Her department at the time of the study, the City of London Migraine Clinic, received funding from Merck for undertaking this clinical trial.
LKM has received research grants and honoraria from Merck, has been a consultant for Merck, and has served on Merck advisory boards and speakers bureau.
WPJVO’s employer, Leiden University Medical Center, received funding from Merck to conduct this clinical trial. WPJVO also received financial support for conference visits from Allergan and Menarini.
MDF’s employer, Leiden University Medical Center, received funding from Merck to conduct this clinical trial.
Acknowledgments
Sheila Erespe from Merck & Co. Inc assisted with formatting of the manuscript. The following investigators participated in the study:
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.