
Abstract
Background
Migraine, particularly with aura, increases the risk for ischemic stroke, at least in a subset of patients. The underlying mechanisms are poorly understood and probably multifactorial.
Methods
We carried out an extended literature review of experimental and clinical evidence supporting the association between migraine and ischemic stroke to identify potential mechanisms that can explain the association.
Results
Observational, imaging and genetic evidence support a link between migraine and ischemic stroke. Based on clinical and experimental data, we propose mechanistic hypotheses to explain the link, such as microembolic triggers of migraine and enhanced sensitivity to ischemic injury in migraineurs.
Discussion
We discuss the possible practical implications of clinical and experimental data, such as aggressive risk factor screening and management, stroke prophylaxis and specific acute stroke management in migraineurs. However, evidence from prospective clinical trials is required before modifying the practice in this patient population.
Introduction
Migraine is the most common neurological disorder and a major cause of disability in the western world, with a prevalence of approximately 13% (9% among men, 18% among women in the United States (US)) (1). An aura is present in up to 30% of migraineurs, usually during the hour preceding the headache (2,3). Spreading depression (SD), an intense neuronal and glial depolarization wave that slowly propagates in brain tissue at a rate of around 3 mm/min, is widely accepted as the electrophysiological substrate of migraine aura (4,5).
Migraine has traditionally been viewed as a benign, chronic episodic condition. However, accumulating evidence suggests that migraine, particularly with aura, can be associated with increased risk for stroke and white matter lesions (6–12). The association is even more striking considering the clinical contrasts between migraine and stroke. Unlike migraine, stroke is an acute and often catastrophic cerebrovascular event. In contrast to the perceived benign nature of migraine (i.e. no imminent risk of injury), stroke is the leading cause of acquired physical disability in adults in the US (13), and the second leading cause of mortality worldwide (14). The prevalence of both ischemic (85% of all strokes) and hemorrhagic stroke in the US is 2.9% in individuals 18 years or older (13), much lower than the prevalence of migraine. And lastly, stroke is predominantly a disease of the elderly, while migraine prevalence peaks around age 40. As per International Headache Society (IHS) criteria, migraine headache may be termed secondary when it is present as part of an underlying disease process such as patent foramen ovale (PFO) or antiphospholipid antibody syndrome; many of these disorders are also associated with increased risk of cardiovascular and cerebrovascular events, discussed in detail below.
In this paper, we briefly summarize the evidence supporting a clinical association between migraine and stroke, propose mechanistic hypotheses that may explain the association, and review the experimental data supporting or refuting some of these hypotheses. Inevitably, in the absence of robust evidence the proposed mechanisms remain speculative. Our overarching aim is to stimulate translational investigations on the mechanisms linking migraine and stroke toward improved patient care. Of note, migraine also appears to increase the risk of hemorrhagic stroke (15). However, available data are less robust, and mechanistic insight is lacking particularly in the experimental setting; therefore, we will limit the discussion to ischemic stroke.
Clinical evidence linking migraine and stroke
Observational data
Although limited by the lack of biomarkers to identify migraine with certainty and quantitative data on intensity, duration and frequency of attacks, abundant observational data from retrospective or population- or hospital-based case-control studies as well as small and large population-based prospective studies including tens of thousands of individuals have firmly established a link between migraine and ischemic stroke, which have been the subject of three meta-analyses (10,16,17). The most recent meta-analysis (10) of 13 case-control and eight cohort studies with a total of 622,381 participants also showed a link between migraine and ischemic stroke with an odds ratio (OR) of 2.04 (95% confidence interval (CI) 1.72–2.43).
The association relied on migraine with aura (OR 2.51, 95% CI 1.52–4.14), and was not significant in migraine without aura (OR 1.29, 95% CI 0.81–2.06). Further subgroup analyses revealed a stronger association in women (OR 2.89, 95% CI 2.43–3.45) (10), as well as in patients younger than 45 (OR 2.65, 95% CI 1.41–4.97), in smokers (OR 9.03, 95% CI 4.22–19.34) and in women using oral contraceptives (OR 7.02, 95% CI 1.51–32.68) (17). Moreover, in the Women’s Health Study (WHS), increased risk appeared to be mainly in those who experienced active migraine attacks within the year before completing the baseline questionnaire, and not in those with just a history of migraine without recent attacks (OR, 1.91 95% CI 1.17–3.10) (18). The risk was also higher in those who experienced >12 attacks per year (OR, 1.7 95% CI 1.1–2.8) in the Stroke Prevention in Young Women study (19). Interestingly, most ischemic events appeared to be transient, or strokes with good clinical outcomes (modified Rankin Scale 0 to 1) in the WHS (20).
Much less is known about the stroke subtype in association with migraine. A tendency for strokes of undetermined cause (OR 1.4, 95% CI 0.9–2.0) or lacunar strokes has been suggested (OR 1.5, 95% CI 0.7–3.3) (19), and in the Italian Project on Stroke in Young adults Study (21) the frequency of right to left shunts was higher in stroke in migraineurs with aura (OR 2.41, 95% CI 1.47–3.95). Based on clinical presentation, a predilection for anterior or posterior circulation has not been demonstrated (19,21).
Lastly, there may be an association between migraine and systemic cardiovascular event risk (e.g. myocardial ischemia and infarction, cardiovascular mortality, peripheral vascular disease). Although an earlier meta-analysis of eight studies did not show an increased risk of myocardial infarction among migraineurs (OR 1.12, 95% CI 0.95–13.2) (17), a more recent large case-control study suggested a higher risk (OR 2.16, 95% CI 1.7–2.76) (22). Most recently, a population-based prospective study showed an increased risk of ischemic heart disease (hazard ratio (HR) 2.5, 95% CI 1.8–3.5) in participants between the ages of 18 and 45 (23). Likewise, a large prospective cohort with a median follow-up of 26 years suggested increased cardiac mortality among migraineurs with aura (24); however, the association was not significant in a meta-analysis (25).
Neuroimaging
A number of neuroimaging studies over the past decade revealed a higher prevalence of subclinical brain abnormalities in migraineurs, including infarcts and white matter hyperintensities, suggesting acute or chronic ischemic disease.
Infarcts
Cerebral Abnormalities in Migraine and Epidemiological Risk Analysis (CAMERA) was a cross-sectional, population-based magnetic resonance imaging (MRI) lesion prevalence study in patients between the ages of 30 and 60 (mean age 48; 161 migraine with aura, 134 migraine without aura and 140 matched controls) randomly selected from the Genetic Epidemiology of Migraine study. Results suggest increased risk of subclinical posterior circulation infarct-like lesions, mostly located in the cerebellum, in migraineurs compared to controls (OR 7.1, 95% CI 0.9–55) (11,26). The risk was substantially higher in migraineurs with aura (OR 13.7, 95% CI 1.7–112), especially with frequent migraine attacks (≥1 attack/month) (OR 15.8, 95% CI 1.8–140), and independent of triptan use or vascular risk factors, although the study was not powered to test the latter. There was no difference in the frequency of such lesions in the anterior circulation. In the nine-year follow-up of the same cohort, none of the lesions disappeared, and new posterior circulation infarct-like lesions were found in 5% of migraineurs compared with none in control subjects (27).
The overall conclusions were later independently confirmed in the Age Gene/Environment Susceptibility Reykjavik study (28), albeit using similar criteria. In this population-based cohort study, 689 patients with a mean age of 60 years (i.e. mid-life) were interviewed for migraine status, and MRI was performed at a mean age of 76 years. This study revealed that women (but not men) who reported active migraine with aura in mid-life had an increased risk of late-life infarct-like lesions compared to non-migraineurs (OR 1.9, 95% CI 1.4–2.6), independently from vascular risk factors. These lesions were also mostly in the cerebellum.
The population-based Epidemiology of Vascular Aging study (780 participants, mean age of 69) also found an increased risk of cerebral infarcts in migraineurs with aura only (29). Although the interpretation was limited by the relatively small number of patients with migraine with aura (17 out of 116 migraineurs), the definition of infarct was stricter, and therefore, data were more specific for ischemic mechanisms. The lesions were mostly located outside the cerebellum and the brain stem.
There have been conflicting data as well, albeit from smaller datasets or from studies designed to test other associations. For example, the Helsinki Young Stroke Registry of 669 patients with first-ever stroke between 15 and 49 years of age did not find any association between the presence of silent brain infarcts and migraine status, despite an overall high frequency of silent brain infarcts (13%) (30). All patients with a lesion had at least two vascular risk factors. Similarly, a cohort of 100 consecutive women with chronic migraine with (51) or without (49) aura (mean age 44) revealed a much lower frequency of infarct-like lesions on MRI than expected by the high frequency of migraine attacks (6%, all with associated vascular risk factors); however, the absence of a control group precluded firm conclusions (31).
White matter hyperintensities
A meta-analysis of retrospective case-control studies (312 subjects, 317 controls) also suggested an increased risk for white matter MRI hyperintensities in migraineurs (OR 3.9, 95% CI 2.26–6.72), even in younger individuals and when controlled for comorbid vascular risk factors (32). Most recent meta-analysis of this association suggested an increased risk in migraine with aura (OR 1.7, 95% CI 1.1–2.7), but not in migraine without aura (OR 1.3, 95% CI 0.96–1.87) (12).
Although the CAMERA study did not show a difference for periventricular or deep white matter lesions between migraineurs and controls, subgroup analysis revealed higher risk for deep white matter lesions only in women with migraine (OR 2.1 95% CI 1.0–4.1); there was no effect of aura status and the risk increased with increasing attack frequency (11). A subsequent analysis also suggested an increased prevalence of infratentorial (mostly pontine) hyperintensities in migraineurs with and without aura (33). The nine-year CAMERA follow-up revealed a higher incidence of deep white matter lesions in women with migraine (OR 2.1 95% CI 1.0–4.1), highest in the migraine without aura group (27). There was no association between lesion progression and attack type, duration, frequency and cumulative number, or specific migraine treatments.
In the Epidemiology of Vascular Aging study, individuals with lifetime history of severe headaches (116 migraineurs and 47 non-migraine headache) were more likely to be in the highest tertile of total white matter hyperintensity volume (OR 2.0, 95% CI 1.3–3.1), preferentially located in deep white matter regions in migraineurs with aura (29). More recently, the prospective Atherosclerosis Risk in Communities cohort of 1028 patients with a mean age of 60 showed an increased risk of moderate to severe white matter MRI hyperintensities (defined as a score ≥3 on a visual rating scale from 0 with no white matter hyperintensity to 9 with confluent and extensive white matter hyperintensities) in migraineurs without aura compared to participants without headache (OR 1.87, 95% CI 1.04-3.37) (34). However, follow-up between eight to 15 years did not reveal a difference in progression of white matter hyperintensities between migraineurs and controls.
In summary, structural brain imaging strongly suggests an increased risk of infarcts and white matter hyperintensities in migraineurs, and perhaps all severe headaches. The conclusions, however, have not been unanimous. The vascular origin of infarcts and white matter hyperintensities has not been confirmed, in part because of imprecise definition of lesions on MRI and lack of neuropathological correlation (35). Furthermore, the association between the severity or duration of migraine and the severity of structural brain abnormalities is inconsistent. An association may be absent, or may be obscured by low power of individual studies and differences in the definition of lesion severity. It should be remembered, however, that migraineurs suffer countless attacks during their lifetime, but develop only a small number of neuropathological lesions. Therefore, it is unlikely that individual attacks directly cause injury.
Monogenic and other rare diseases
Albeit rare, monogenic diseases may help understand the pathophysiology of more common polygenic or multifactorial conditions. An association between migraine and stroke is further supported by frequent co-existence of the two in monogenic diseases. For example, migraine with aura is often the first symptom in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), present in 20%–40% of patients. CADASIL is the most common monogenic inherited cerebral small-vessel disease in middle-aged adults. It is caused by mutations in the NOTCH3 gene that are exclusively expressed in vascular smooth muscle cells in the adult brain. Besides migraine with aura, CADASIL patients progressively develop subcortical infarcts, mood disorders and cognitive impairment culminating in frank dementia (36). Most migraine attacks are typical, but 50% of patients also experience attacks with atypical aura including basilar or hemiplegic migraines, or prolonged migraine auras (37).
Hereditary infantile hemiparesis, retinal arteriolar tortuosity and leukoencephalopathy (HIHRATL) is another autosomal dominant cerebral small-artery disease associated with migraine with or without aura. Mutations in the COL4A1 gene encoding type IV collagen α1 chain destabilize the triple helix domain of collagen IV found in the basement membranes. First described in mutant mice with porencephaly generated by random mutagenesis (38), COL4A1 mutations have now been detected in many human families, in whom newborns and adults may be affected (39,40). The phenotype is variable, and besides migraine, may include infantile hemiparesis, porencephaly, seizures, white matter hyperintensities, hemorrhagic more than ischemic stroke, renal and ocular vessel tortuosity, and intracranial asymptomatic aneurysms (41).
Retinal vasculopathy with cerebral leukodystrophy is an autosomal dominant small-vessel disease of middle-age onset, previously known as cerebro-retinal vasculopathy, hereditary vascular retinopathy and hereditary endotheliopathy, retinopathy, nephropathy and stroke. All entities have been linked to mutations in the TREX1 gene (42), which encodes a DNA-specific exonuclease implicated in DNA repair under conditions of oxidative stress. The clinical presentation includes progressive loss of visual acuity related to retinal vasculopathy, small cerebral infarcts and white matter hyperintensities that can coalesce to form pseudotumors. Although migraine or “migraine-like” headache has been described in all phenotypes, migraine, equally with or without aura, seems to be more frequent in hereditary vascular retinopathy (43), where it is found in 70% of patients and often associated with Raynaud phenomenon.
Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) is caused by mutations in the mitochondrial genome with a maternal pattern of inheritance. It mostly affects children and young adults, with 90% of patients having symptoms before 30 years of age. Attacks of migraine are present in 70% of cases and may be isolated, or more frequently accompany seizures and/or focal or global neurological deficits related to post-ictal symptoms or ischemic stroke. Remarkably, strokes occur early in life, do not match a particular vascular territory, often progress slowly (over days or weeks) and can often be partially reversible, which justifies the term “stroke-like episodes.” However, residual deficits often accumulate leading to motor, visual or cognitive disability. Systemic features include short stature, sensorineural hearing loss, diabetes mellitus, cardiac disease, myopathy, and gastrointestinal and renal involvement (44–46).
Hereditary hemorrhagic telangiectasia is an autosomal dominant disorder characterized by mucocutaneous telangiectasia and arteriovenous malformations of the brain, lung, gastrointestinal tract and liver due to mutations of endoglin or activin receptor-like kinase 1 (47). Migraine is present in up to 40% of patients, in addition to the classical epistaxis and other sites of hemorrhage that start almost always before 40 years of age. Although migraine and stroke, mostly hemorrhagic, may be related to intracerebral arteriovenous malformations (48), migraine, mostly with aura, occurs typically independent of cerebral vascular malformations and is associated with an increased probability of pulmonary arteriovenous malformation, which is also associated with an increased risk of cerebral ischemic events (49–51). Interestingly, in hereditary hemorrhagic telangiectasia patients without a pulmonary arteriovenous malformation, prevalence of migraine without aura was also higher in one study (52). Outside of hereditary hemorrhagic telangiectasia, an association between migraine and occipital arteriovenous malformations has been noted (53), and migraine is a recognized accompaniment of Sturge-Weber syndrome (54), and has been noted in association with moyamoya disease (55), underscoring the link between vascular structural abnormalities and migraine.
Potential caveats in interpretation of clinical evidence
Heterogeneity of migraine and inconsistent use of diagnostic criteria, referral bias, difficulty differentiating between a migraine attack and a transient ischemic attack (TIA) in the absence of biomarkers, presence of comorbidities and concurrent medications, and overrepresentation of younger patients can all complicate and confound the interpretation of clinical data. It is highly unlikely that increased stroke risk in migraine with aura can be explained entirely by secondary migraine in the setting of other diseases such as CADASIL or hereditary hemorrhagic telangiectasia. These potential caveats notwithstanding, the evidence firmly supports an association, and in many cases provides clues to the pathogenesis.
Mechanisms of association
Observational, genetic and neuroimaging data suggest that migraineurs with aura have an increased risk of ischemic stroke. The nature and mechanisms of elevated risk and whether the risk is modifiable are unclear, and the risk likely applies only to a subset of migraineurs. For obvious reasons, we cannot perform experiments to dissect the mechanisms linking migraine and stroke in human subjects, and one has to employ experimental animal models to test hypotheses.
Based on available clinical and experimental data, one can postulate mechanisms to explain the association between migraine and ischemic stroke. The conceptual framework (Figure 1) is undeniably an oversimplification, but nevertheless allows one to generate hypotheses. These mechanisms are not mutually exclusive; they may be operational in different patients, or overlap in the same patient and interact to synergize. Some of the potential mechanisms are supported by clinical and experimental data, which we will discuss individually and elaborate on in specific subcontexts.
A conceptual framework for possible mechanisms linking migraine and stroke based on available direct or indirect clinical and experimental evidence. (a) Upstream vascular and metabolic risk factors for cerebral ischemia (e.g. endothelial dysfunction, cervical artery dissection, hypercoagulable states, paradoxical embolism, adverse lifestyle) can trigger migraine with aura when mild, and cause ischemic stroke when severe. (b) Upstream genetic risk factors that enhance susceptibility to spreading depression (SD) as a migraine trigger may sensitize the tissue to ischemia, so that infarction ensues with a milder degree of ischemia or ischemic injury progresses more rapidly. (c) Migraine with aura may increase vascular and metabolic risk of ischemia (e.g. migrainous infarction, vasoconstrictive complications of migraine medications, adverse lifestyle, endothelial dysfunction or platelet aggregability caused by migraine with aura).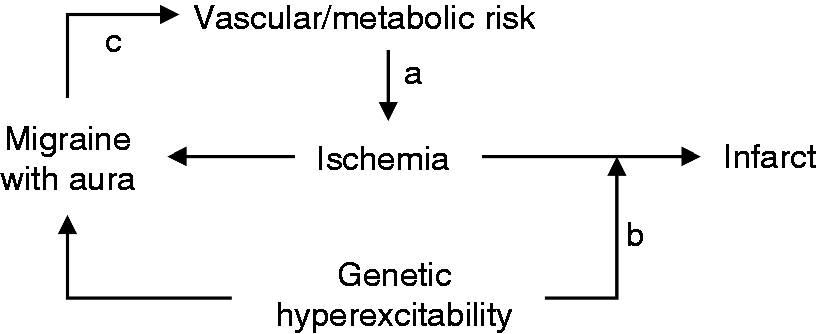
Migrainous infarction
A first report of migrainous infarction goes back to the 19th century (56). As defined by IHS, migrainous infarct is an ischemic infarct demonstrated by neuroimaging that develops during a typical migraine with aura attack with abnormally prolonged aura symptoms (>60 min), in a patient with a history of migraine with aura, in the absence of an alternative cause for the infarct (2). Implicit in this definition is the fact that migrainous infarct is caused by the migraine attack. Therefore, migrainous infarction is by definition a diagnosis of exclusion, and depends on the depth of work-up for an alternative etiology.
The rarity (∼0.5% of all ischemic strokes) of true migrainous infarction is further underscored in clinical series. A retrospective multicenter study spanning more than 20 years identified only 33 patients who fit the IHS criteria for migrainous infarct, almost half of the patients had no other vascular work-up than Doppler ultrasonography, and more than a third did not have transoesophageal echocardiography (57). Moreover, 40% of patients indeed had a PFO. In a prospective study spanning 11 years, migrainous infarcts made up only 0.2% of all etiologic diagnoses; a third of migrainous infarcts was a first-ever aura attack (i.e. did not fulfill the IHS criteria), two-thirds had a PFO, and almost all had other vascular risk factors (58). Moreover, detailed investigations revealed rare causes of stroke in almost a quarter of those originally diagnosed as migrainous infarction.
The diagnosis has become even more problematic after clinical and experimental evidence clearly showed that ischemic events can trigger symptomatic migraine attacks, as well as a SD as the aura surrogate (59–62). Hence, a primary ischemic event can cause prolonged neurological signs and symptoms that may be perceived as aura; therefore, migrainous infarct should not be diagnosed without an extensive work-up to exclude other causes. The condition is even more likely to be over-diagnosed in young patients, as 40% of ischemic strokes in the young remain cryptogenic.
Nevertheless, there may be a mechanistic basis for migraine aura culminating in infarction. SD (i.e. aura) has profound metabolic and hemodynamic effects on the brain tissue. While adenosine triphosphate (ATP), O2 and glucose consumption increase dramatically during and after SD, substrate delivery via blood flow may not be sufficient, thereby creating supply-demand mismatch (63–65). Although SD by itself is not injurious in otherwise healthy brain tissue (66), under certain conditions, SD may be associated with abnormal neurovascular coupling, causing marked and prolonged vasoconstriction (i.e. inverse coupling) and severely limiting tissue O2 delivery (67,68). For example, reduced nitric oxide levels when combined with mildly elevated extracellular K+ or low glucose leads to constriction and laminar cortical necrosis during SD (69,70). In theory, such ionic disturbances may even be precipitated by endothelial ion channel or pump dysfunction (71). However, whether genetic, hormonal or environmental factors can modulate the hemodynamic and metabolic response and create severe supply-demand mismatch during SD to precipitate infarction in otherwise healthy brain remains to be proven. At least in MELAS, such a mechanism is possible (see below).
Migraine drugs predisposing to ischemic events
Another potential mechanism one can postulate to explain increased stroke risk in migraineurs is exposure to high-risk external factors linked to migraine. For example, drugs frequently used in the management of migraine may increase stroke risk. Indeed, triptans and ergotamine have vasoconstrictive properties, and are not recommended in patients with complicated migraine with aura due to the associated oligemia. Moreover, these drugs, as well as serotonin reuptake inhibitor antidepressants, can precipitate reversible cerebral vasoconstriction syndrome (RCVS), a clinical and radiological syndrome characterized by severe unusual headache and transient multifocal cerebral vasoconstriction that can lead to stroke, subarachnoid hemorrhage and/or brain edema (72). Interestingly, migraine is more common in RCVS patients even in the absence of an offending drug treatment (72), suggesting that other factors also play a role.
However, large studies have not substantiated a link between triptan use and the risk of stroke in migraineurs (e.g. General Practice Research database, HR 1.13, 95% CI 0.78–1.65; United Healthcare database, OR 0.90, 95% CI 0.64–1.26) (73,74). A large, nested case-control study of cardiovascular events in triptan overuse also did not show a significant increase in the risk of hospitalization for ischemic events (OR 1.43, 95% CI 0.82–2.49) (75). And lastly, MR angiography has recently failed to show vasoconstriction in intracranial arteries upon triptan administration, despite vasoconstriction of extracranial vessels (76). Ergotamine did increase the risk of hospitalization for cardiovascular and cerebral ischemic events (OR 2.55, 95% CI 1.22–5.36); however, this occurred only in patients with other cardiovascular comorbidities who overused the drug (75), and was not confirmed in other studies (OR 1.49, 95% CI 0.93–2.41) (74). Moreover, ergotamine has not been a commonly used drug clinically. It should also be noted that the association between migraine and stroke is mainly driven by migraine with aura, while migraine drugs are used by all migraineurs regardless of aura status. For these reasons, migraine drug use is unlikely to explain the increased stroke risk in migraine, unless there is a specific interaction with aura.
Migraine predisposing to ischemic events indirectly through lifestyle changes
Migraine can have profound effects on a patient’s mood and lifestyle, and may indirectly augment behavior patterns posing a vascular risk. For example, obesity has been associated both with episodic and chronic migraine, mostly in patients with high attack frequency (77–79), although the direction of causality is not clear. Migraine could predispose to obesity, as women with a history of childhood migraine have a higher risk of weight gain later in life (80). In theory, obesity may be a side effect of migraine treatments, or due to reduced physical activity to avoid triggering or exacerbating migraine attacks, although regular exercise could also reduce migraine attacks (81). Indeed, migraineurs also exercise less, as yet another factor that can increase cardiovascular risk (82). Conversely, obesity may predispose to migraine through hormones released from the adipose tissue, such as adiponectin (83). Finally, migraine and obesity may also be linked by a shared underlying disorder, such as abnormalities in hypothalamic function or serotonergic transmission (84). Regardless of the causal relationship, obesity and associated metabolic syndrome are well-known stroke risk factors, and may also link migraine with coronary and peripheral artery disease (17,22,85).
Indeed, the Genetic Epidemiology in Migraine study, a population-based, cross-sectional study including 620 migraineurs and 5135 non-migraineurs, showed that patients with migraine, in particular with aura, are more likely to smoke and have unfavorable cholesterol profiles and higher Framingham risk scores than non-migraineurs (86). This was corroborated by the American Migraine Prevalence and Prevention study, a case-control study including 6102 patients and 5243 controls, which showed that migraineurs were more likely to have vascular risk factors and higher Framingham scores (22). The lower consumption of alcohol by migraineurs (82,87), probably avoidance due to its ability to induce attacks, could also increase stroke risk in migraineurs by the loss of its vascular protective effect when consumed in moderate amounts. Moreover, migraineurs are more likely to smoke (82,86), or use hormone therapy after menopause (29), as additional risk factors. Lastly, migraine and depression are linked, and although bidirectional causality has been suggested (88), recent data from the WHS suggest that migraine (as well as non-migraine headache) increases the risk of incident depression in middle-aged women (relative risk 1.48, 95% CI 1.37–1.60), particularly in individuals with high attack frequency (89). Depression is a risk factor for stroke (90), and may contribute to increased stroke risk in migraineurs. Based on these data, one might speculate that often-modifiable lifestyle factors may add to increased stroke risk in migraineurs.
However, large-scale epidemiological studies have also shown that stroke risk in migraineurs is independent of vascular risk factors (10,16,17). For example, the WHS revealed that the association between active migraine with aura and ischemic stroke was apparent only among women in the lowest Framingham risk score group (87). Likewise, the Italian Project on Stroke in Young adults Study showed lower vascular risk factor profile in migraineurs with stroke, and the Stroke Prevention in Young Woman study showed that women without a history of hypertension, diabetes or myocardial infarct were at greatest risk of migraine-associated stroke (19,21). Moreover, large-artery atherosclerosis as a stroke mechanism did not differ between migraineurs and controls. Lastly, no correlation was found between migraine status and atherosclerosis markers (e.g. intima-media thickness, pulse wave velocity and ankle-brachial index) in a large case-control study including 617 controls and 360 migraineurs (91), further arguing against a role for atherosclerosis as the link between migraine and stroke. Unfortunately, there are no experimental data to support or refute these potential mechanisms.
Migraine and cervical artery dissection
Cervical artery dissection is a well-known cause of stroke, particularly in the young population. A meta-analysis of case-control studies has shown that migraineurs have an increased risk of cervical artery dissection with an OR of 2.06 (95% CI 1.33–3.19) (92), later confirmed in a larger international multicenter study consisting of 968 stroke patients with dissection and 653 stroke patients without dissection (Cervical Artery Dissection and Ischemic Stroke Patients study; OR of 1.51, 95% CI 1.15–1.99) (93). In a recent series, 60% of patients who presented with concurrent cervical artery dissection and reversible cerebral vasoconstriction syndrome were migraineurs (94). The mechanisms responsible for cervical artery dissection are poorly understood (95), as is the causal relationship with migraine. Of course, ischemia due to embolism or hemodynamic insufficiency precipitated by a dissection can directly trigger a migraine attack (59), as well as stroke. In this context, migraine attacks can be considered TIAs. It may be speculated that endothelial injury and microdissections are not infrequent in everyday life and most go unnoticed. A small hemodynamically insignificant dissection invisible by routine imaging tools can still activate platelet aggregation and serotonin release, which may increase the likelihood of a migraine attack without causing stroke (96). However, absence of a predilection for migraine with aura is not fully congruous with an ischemic trigger for migraine in this setting. Conversely, migraine may increase the risk for cervical artery dissection, for example, by inducing matrix metalloproteinase (MMP) expression (97), the levels of which are elevated in migraineurs both ictally and interictally (98), and in patients with cervical artery dissection (99). Indeed, a significant association has been demonstrated between migraine, especially with aura, and interictal serum elastase activity (100). Therefore, chronic (i.e. ictal and interictal) elevation of MMP and elastase levels in migraineurs may perhaps cumulatively weaken the arterial wall to predispose to dissections upon mild traumatic insults. Although unrelated to dissections, a curious relationship has been reported between anatomical variations in the posterior circle of Willis (i.e. fetal posterior cerebral artery origin or basilar hypoplasia) and migraine with or without aura (101). Although this is an interesting concept, the data have been inconsistent among studies and remain to be replicated (102,103).
Hypercoagulability and endothelial dysfunction
Endothelial dysfunction has also been suggested as a possible link between migraine and stroke (104). Studies of vascular reactivity as a surrogate for endothelial function in migraineurs have revealed conflicting results. In peripheral arteries vascular reactivity was reported decreased (105–107) or unchanged (108–111) in different studies. Similarly, cerebrovascular reactivity was either increased (112–114) or decreased (115–117). Further complicating the issue, cerebral and peripheral vascular reactivity do not always correlate (109), anterior and posterior circulation often differ in reactivity readouts (118), and subject selection (e.g. aura status, exclusion of vascular comorbidities) and the method used to assess reactivity are highly variable among studies. Biomarker studies also suggest endothelial dysfunction. Migraineurs have higher interictal levels of circulating t-PA, high-sensitivity C-reactive protein, von Willebrand factor, vascular endothelial growth factor and nitric oxide metabolites, some only in migraineurs with aura (110,119). Likewise, the number of circulating endothelial progenitors cells, believed to repair injured endothelium, is lower in migraineurs (110,120). It is not known whether migraine and stroke are both facilitated by the underlying endothelial dysfunction, or whether migraine can facilitate endothelial dysfunction as a stroke risk factor. Overall, the clinical relevance of endothelial dysfunction and its role linking migraine and ischemic stroke, if any, remain to be tested. The association between migraine and livedo reticularis (e.g. Sneddon’s syndrome) is interesting to note in the context of vascular endothelial dysfunction (121).
Data on acquired or inheritable hypercoagulable states in migraineurs have also been conflicting and not always replicated. In a small early study consisting of 35 migraineurs and 24 controls, elevated prothrombin fragment 1.2 levels in migraineurs with aura suggested activation of clotting cascade (122). Some studies suggested a hypercoagulable state in migraineurs (123,124), while others failed to show a difference from controls (125–129). Low sample size, varying from 20 to 276 participants, has been a major limitation. Studies on specific markers of acquired or inherited hypercoaguble states have been more conclusive. A recent study in 1456 women (mean age 34) with a personal or familial history of venous thrombosis has shown an increased risk of migraine with aura in carriers of factor V Leiden or factor II G20210A mutations (OR 1.76, 95% CI 1.02–3.06), strongly supporting an association between thrombophilia and migraine (130). Moreover, a higher prevalence of hypercoaguble state was found in stroke patients with migraine when compared to non-migraineurs, providing further support to the association (21,131). Other causes of hypercoagulable states, such as antiphospholipid syndrome, systemic lupus erythematosus and polycythemia, have also been linked to migraine with aura in case-control studies, small case series and anecdotal reports; however, in the absence of robust datasets, some of these associations may be spurious (132–134). Lastly, data from two studies consistently showed that methylenetetrahydrofolate reductase (MTHFR) 677 TT genotype combined with migraine aura confers an increased risk of ischemic stroke (OR 1.81, 95% CI 1.02–3.22, and HR 4.19, 95% CI 1.38–12.74) (135,136). The MTHFR TT genotype appears to increase the risk of migraine with aura, whereas angiotensin-converting enzyme II genotype decreases the risk of any migraine (137), as other potential shared genetic modulators of stroke risk.
Paradoxical embolism predisposing to migraine and ischemic events
PFO is an incomplete closure of the fetal communication between right and left atrium that can serve as a conduit for circulating particulate and chemical substances to bypass pulmonary circulation, and reach the brain unfiltered. Classical epidemiological data have linked migraine and PFO. A meta-analysis of case-control studies have demonstrated an increased risk for migraine in patients with PFO (OR 5.13, 95% CI 4.67–5.59) and an increased risk for PFO in migraineurs (2.54, 95% CI 2.01–3.08); the association is even stronger in migraine with aura (138). Furthermore, a study in 20 families with PFO and migraine with aura has suggested an autosomal pattern of inheritance of atrial shunts that in some families could be linked to inheritance of migraine with aura (139). Highly promising anecdotal data from retrospective open-label studies on the efficacy of PFO closure in reducing migraine attack frequency, such as headache resolution in up to 80% of patients (138), particularly in patients who experience a migraine attack during PFO closure (140), paved the way for the Migraine Intervention with STARFlex Technology trial (MIST). MIST randomized 147 migraineurs with aura and a PFO with moderate to large right-to-left interatrial shunt, to PFO closure or sham procedure in a double-blind fashion. The primary and most secondary outcome measures did not differ between treatment and sham arms, effectively MISTifying the field, and hampering forward progress. However, the MIST trial included only patients with highly frequent attacks (∼5 attacks and five to 23 headache days/month) who had previously failed at least two prophylactic treatments (141). Hence, most patients had intractable migraines unresponsive to multiple therapeutic trials; therefore, inclusion criteria were chosen not as an indication but a justification for an invasive procedure. Although this was probably not the best cohort in which to test the efficacy of an experimental intervention, the results of a recent meta-analysis of well-designed, unbiased studies also questioned the existence and strength of an association between migraine and PFO (142).
Nevertheless, PFO is clearly linked to cryptogenic stroke (143), and may act as a source of arterial microemboli or chemical offenders. For example, injection of sclerosing agents to treat varicose veins triggered a migraine with aura attack in a subset of individuals, almost all of whom harbor a PFO (144,145). Moreover, intravenous injection of agitated saline with air microbubbles has been reported as a migraine trigger in patients with a PFO and large right-to-left shunt (146,147). Besides triggering an attack, microbubbles can also alter cerebral electrical activity in migraineurs with aura and a PFO, but not in migraineurs with aura without a PFO or in patients with a PFO but no migraine history (148), perhaps suggesting that microvascular hypoxia/ischemia may induce cortical SD in a susceptible set of patients.
Indeed, the principle that microemboli can trigger SD as an aura surrogate without causing ischemic injury has been tested and proven in the experimental setting. Intracarotid infusion of particulate material of various size and compositions (e.g. cholesterol particles of <70 µm, polystyrene microspheres of 5–20 µm), as well as air microbubbles (10 µl), reliably evoked cortical SD in mice (60). The mechanism involved transient cerebral hypoperfusion as shown by real-time, full-field blood flow imaging using laser speckle flowmetry. The probability of SD induction related to the duration and severity of hypoperfusion, which in turn was determined by the size and composition of the emboli. When microembolic hypoperfusion reached a threshold severity and duration, an SD occurred. In more than half the animals that developed an SD, no ischemic injury was detected by a meticulous examination of serial histological sections throughout the brain, as well as by MRI. This study suggests that in a subset of migraineurs, transient cerebral ischemia induced by microemboli may be responsible for triggering a migraine attack, and when ischemia is severe enough, triggering an ischemic stroke. In humans, PFO, as well as pulmonary arteriovenous malformation such as in hereditary hemorrhagic telangiectasia, and atrial myxoma (149), may serve as a source of microembolism. Of course, microembolism is unlikely to be the trigger for every attack in all migraineurs with aura and a right-to-left shunt.
Right-to-left intracardiac shunts may also allow neuroactive and/or vasoactive chemicals to access the brain. One such chemical is endothelin, plasma levels of which are elevated during a migraine attack (150–153). Interestingly, endothelin A receptor polymorphisms have been shown to modulate migraine risk (154). Although air microembolism has been hypothesized as a mechanism for attacks of migraine often with aura after foam sclerotherapy of varicosities (144,145), attacks were also triggered after liquid sclerotherapy, which does not predispose to air embolism. An alternative hypothesis is endothelin release from the irritated endothelium during sclerotherapy into the venous circulation gaining access to the brain through a right to left shunt (155). This is supported by elevated plasma endothelin concentrations after sclerotherapy in rats, regardless of whether liquid or foam sclerosing agent was used (155,156). Similar plasma endothelin elevations have also been shown in humans after foam sclerotherapy (156). Endothelin 1 indeed potently triggers SD in rats through endothelin A receptors (157,158), and the mechanism involves severe vasoconstriction and hypoperfusion, leading to infarction, providing a direct link between migraine and ischemic stroke (159). Altogether, these data suggest that right-to-left shunts are associated with migraine and can explain a subset of ischemic strokes in migraineurs.
Increased sensitivity to ischemic injury
An alternative hypothesis to explain a migraine-stroke association is that cerebral hyperexcitability phenotype associated with migraine sensitizes the tissue to ischemia. Recent experimental data in mice expressing familial hemiplegic migraine (FHM) type 1 mutations provided direct support for such a mechanism. FHM is an autosomal-dominant subtype of migraine with often severe and prolonged auras associated with motor deficits, sometimes accompanied by sensory, aphasic, visual and basilar symptoms. A third of patients can experience a decrease in level of consciousness and even coma, which may be prolonged (160). FHM has been used as a model for more common forms of migraine with aura because of shared clinical features and trigger factors, female preponderance, and because two-thirds of FHM patients and their first-degree relatives also suffer from attacks of common migraine with and without aura.
FHM type 1 is caused by mutations in the pore-forming α1A subunit of Cav2.1 voltage-gated Ca2+ channels, which are critical for presynaptic glutamate release. Mutant channels open on smaller membrane depolarizations and stay open longer. The net result is increased presynaptic Ca2+ entry and glutamate release, resulting in enhanced cerebral excitability (161), a mechanism likely shared with more common forms of migraine (162). Indeed, FHM1 mutants are highly susceptible to SD and frequently develop subcortical and re-entrant SDs with prolonged neurological deficits mimicking those observed in FHM patients (163,164).
Availability of transgenic mouse models expressing FHM1 mutations has recently allowed testing of the hypothesis that the cerebral hyperexcitability phenotype associated with migraine sensitizes the brain tissue to ischemia. In support of the hypothesis, two FHM1 mutant mouse strains developed larger infarcts compared with wild type after transient focal cerebral ischemia (165). The phenotype correlated well with the strength of gain-of-function of each mutation (S218L mutant more severe than R192Q), and showed an allele-dosage effect whereby homozygous mutants fared worse than the heterozygotes. The mechanism was indeed linked to hyperexcitability, because anoxic depolarization developed faster and diffusion-weighted MRI lesions grew rapidly after stroke onset in the mutants. Perhaps more important, mutants developed many more peri-infarct depolarizations (PIDs) during acute stroke. PIDs are SD waves spontaneously triggered within the ischemic penumbra, and expand the infarct by worsening the supply-demand mismatch, thereby explaining the accelerated infarct growth in migraine mutants, as well as in humans (61,166). The data also showed that migraine mutants had an elevated minimum cerebral blood flow threshold required for tissue survival, directly supporting the hypothesis that brain tissue in migraineurs is more susceptible to ischemic injury. The study also showed that female mutants, which are even more hyperexcitable and susceptible to SD than males, developed larger infarcts and worse outcomes after ischemic stroke than wild-type controls, consistent with the clinical observations in women with migraine. Lastly, ischemic brain swelling appeared to be more severe in mutants and could explain the higher mortality in the S218L mutant strain (165).
Altogether, these data suggested that familial migraine mutations (e.g. FHM1) that are known to augment cerebral excitability also facilitate infarction if and when the tissue becomes ischemic, by predisposing to frequent ischemic depolarizations. Of course, whether the mechanism is valid in other monogenic migraine disorders or in sporadic migraine remains to be tested. The hyperexcitability phenotype in migraine appears to also enhance susceptibility to SD, which is likely the final common mechanism for tissue sensitization to ischemia. In this context, it is notable that CADASIL mutant mice (Notch3R90C) as well as Notch3 null mutants show markedly enhanced SD susceptibility (167). Indeed, both Notch3 null mice and transgenic mice expressing CADASIL mutations develop larger infarcts, and at least in the null mutants, this is associated with an increased frequency of PIDs (168,169). Although it is not clear how the mutations in the NOTCH3 gene, which is exclusively expressed in vascular smooth muscle cells in the adult brain, lead to cerebral hyperexcitability, the data strongly support the notion that enhanced SD susceptibility translates into susceptibility to ischemic infarction, which is consistent with the stronger epidemiologic association observed between stroke and migraine with aura compared with without aura. It is important to note that aside from the hyperexcitability, CADASIL mutations clearly lead to cerebral small-vessel disease and vascular dysfunction (36), and increase the risk of occurrence of ischemic events and chronic cerebral hypoperfusion (e.g., lacunar infarcts, white matter disease), in addition to the risk of developing infarction during those events. In other words, mechanisms leading to lacunar infarcts and white matter disease may be different from those leading to hyperexcitability. This is further supported by the observation that the onset of migraine with aura attacks often precedes the emergence of clinical and radiological evidence of ischemic vascular disease in CADASIL, and in fact, attacks usually diminish in frequency and disappear as the ischemic lesion load increases. It remains to be tested whether other mutations clinically associated with small-vessel disease and migraine, such as TREX1 and COL4A1, also augment SD susceptibility and sensitize the brain tissue to ischemic injury. Transgenic mice expressing mutations in these genes have been developed, and at least in case of COL4A1, mutants develop ultrastructural changes in cerebral vasculature, endothelial and smooth muscle cell dysfunction, and blood pressure regulation (170).
And lastly, MELAS is another disease in which migraine and stroke-like episodes coexist on a spectrum. Impaired mitochondrial oxidative phosphorylation in MELAS creates a chronic state of energy shortage and inability to match increased demand, proposed as a potential mechanism linking hyperexcitability and sensitivity to ischemic injury (171,172). Other mechanisms may also be involved, such as accumulation of dysfunctional mitochondria in endothelium and smooth muscle cells of small cerebral vessels leading to endothelial dysfunction and increased capillary permeability (173,174). In the absence of representative animal models, whether the hyperexcitability is in any way related to SD remains to be tested. One can perhaps speculate that inadequate mitochondrial response to increased energy demand during SD impairs SD recovery, and markedly prolongs the depolarization predisposing to tissue injury.
Clinical implications
Migraine as a symptom of TIA: Angina cephalis?
As noted above, clinical and experimental evidence suggests that migraine attacks can be triggered by transient cerebral ischemic events (i.e. symptomatic migraine), and as such, they may carry a similarly elevated risk of impending stroke and should perhaps be treated as a TIA.
Is antithrombotic stroke prophylaxis indicated in migraineurs?
If migraine with aura is a TIA, antithrombotic use may not only diminish the stroke risk but also reduce the frequency of migraine attacks. A similar argument can be made for PFO closure, or for inhibitors of potential vasoactive or neuroactive mediators presumed to pass through a PFO into arterial circulation unfiltered by the pulmonary circulation and predispose to migraine attacks (e.g. endothelin). Although, failure to meet the primary efficacy endpoints in the MIST trial rendered PFO closure in migraine highly controversial, for reasons discussed above (i.e. not the best cohort in which to test an investigational intervention, primary endpoints difficult to meet), we believe the evidence is not sufficient to support or refute the hypothesis at this time. The critical challenge will be to identify the best closure candidates in whom the PFO is symptomatic.
Is aggressive avoidance of exacerbating factors (e.g. oral contraceptive use, smoking, risk of traumatic dissection) indicated in migraineurs?
Along the same lines of reasoning as above, one might argue that given the geometrically increased risk of stroke by the presence of aggravating factors in highly susceptible migraineurs, such factors should be a part of aggressive risk management. For example, the World Health Organization recommends women with migraine with aura avoid combination oral contraceptives. Perhaps the warning against chiropractic cervical manipulations (175,176) should be particularly strong in migraineurs. Once again, reduced risk for ischemic events as a migraine trigger may also decrease the frequency of migraine attacks.
Is complete screening for vascular risk factors (e.g. hypercoaguble state, PFO, genetic mutations) indicated in migraineurs in the absence of a stroke?
Migraine with aura as a potential TIA, particularly when the diagnosis of migraine does not fulfill IHS criteria (e.g. first attack, atypical aura, abnormal neurological exam), when the aura is always on the same side, and in late-onset migraines or when the attack frequency or characteristics changes, might also justify searching for common stroke etiologies as part of the clinical work-up. The approach may also affect migraine classification.
Migraine as a hyperexcitable state that sensitizes the brain to ischemic injury: The tissue factor
The clinical and experimental evidence reviewed above suggests that migraine with aura reflects a hyperexcitable state that enhances susceptibility to SD as a final common mechanism. Experimental evidence also suggests that susceptibility to develop SD is related to the susceptibility to develop injury if and when the brain tissue becomes ischemic. Because aura (perceived or not) is the relevant risk biomarker, the frequency of attacks with aura, rather than the total frequency of attacks only rarely accompanied by aura, appears to be the critical determinant. It should be noted, however, that data supporting a direct link between SD susceptibility and injury susceptibility come exclusively from transgenic animal models of monogenic, familial forms of human migraine, and whether the principle applies to non-familial forms of migraine is not known.
Is therapeutic time window for revascularization in ischemic stroke shorter in migraineurs?
In FHM1 mutant mice, the diffusion-weighted MRI lesion (i.e. ischemic core) expanded rapidly because of enhanced susceptibility to anoxic depolarization and PIDs (165). If hyperexcitable brain tissue in migraineurs readily develops ischemic depolarizations, then the core expands into the perfusion defect leading to rapid loss of viable tissue at risk (i.e. penumbra). This would effectively shorten the therapeutic time window of efficacy for stroke therapies in migraineurs.
Does migraine prophylaxis diminish the stroke or white matter lesion risk or severity in migraineurs?
Migraine prophylaxis suppresses SD susceptibility (177), and therefore, may also diminish susceptibility to ischemic injury by rendering the tissue more resistant to ischemic depolarizations. This approach to reduce tissue sensitivity to ischemia could also be efficacious in non-migraineurs as well.
Is there a clinicopathological disconnect during acute stroke in migraineurs?
PIDs are indistinguishable from SD waves when they propagate into the non-ischemic brain tissue, but they nevertheless cause transient electrophysiological silence in the tissue they invade, and can cause neurological deficits much like the aura symptoms during migraine attacks. If migraineurs develop more frequent PIDs, they may exhibit more severe and perhaps fluctuating neurological deficits than what would be expected based on the size and location of the ischemic lesion as seen on MRI. The concept may be true in other brain injury states as well, such as subarachnoid or intracerebral hemorrhage, and trauma, in which injury depolarizations akin to SD are known to occur in the human brain.
Is malignant brain edema more frequent in migraineurs?
It has been shown that SD disrupts the blood-brain barrier (97). Hence, frequent PIDs may be associated with more severe blood-brain barrier breakdown and ischemic edema formation after stroke, which can even become life threatening in case of large middle cerebral artery or cerebellar infarcts. If so, early decompressive craniectomy may be indicated in migraineurs with aura.
Concluding remarks
Migraine is a stroke risk factor with an effect size and prevalence comparable to other risk factors. Its recognition as an important and perhaps modifiable vascular risk factor, sought and documented as part of the patient’s medical history, will certainly be facilitated by better definition of the mechanisms of this association and the causal relationships. Unfortunately, current diagnostic criteria do not distinguish patients with occasional vs. frequent auras (2), a distinction that is relevant for stroke risk and should be documented. With increased awareness, management of migraine will become more than management of a headache disorder, and more holistic as with other vascular risk factors. Of course, enough evidence to change practice can be achieved only in large prospective clinical studies targeting mechanisms and in therapeutic trials.
Clinical implications
Observational, neuroimaging and genetic data strongly support an association between migraine, particularly with aura, and stroke. Clinical and experimental evidence suggest multiple independent mechanisms. Cerebral ischemic events can trigger migraine, and conversely, migraine can be a marker of increased sensitivity to ischemic injury.
Footnotes
Funding
This work was indirectly supported by grants from the US National Institutes of Health (NIH) (NS055104, NS061505); the American Heart Association (AHA) (11SDG7600037); Fondation Leducq; the Heitman Foundation; the Ellison Foundation; Institut Servier; Philippe Foundation, Thérèse and René Planiol Foundation for the Study of the Brain; and the Association CADASIL France.
Conflict of interest
None declared.