
Abstract
Introduction
Familial hemiplegic migraine (FHM) is a rare subtype of migraine with transient hemiplegic aura.
Patients and methods
We describe three unrelated families with familial hemiplegic migraine type II (FHM2). Retrospectively, information on 47 family members could be obtained, 15 by personal examination and 32 by indirect anamnesis from relatives. Genetic analyses were performed in 13 patients.
Results
One family had a novel missense mutation in the ATP1A2 gene (c.659C>T, p.Ser220Leu) that segregated with the phenotype in three generations. Two further unrelated families with different ethnic backgrounds (one from Germany and one from Russia) had a missense mutation that has not been described as yet in FHM, but occurred in only a single patient with sporadic hemiplegic migraine (c.2723G>A, p.Arg908Gln). Clinically the patients had severe attacks lasting up to several weeks as well as epileptic seizures. Three patients with a proven mutation in the ATP1A2 gene clinically presented without hemiparesis. Furthermore, there was a possible relation of FHM2 to mental retardation in another two patients.
Conclusion
Clinical symptoms may last for several weeks in some patients. Patients with FHM2 may also present without hemiplegia. Therefore, the full family history has to be taken into account to establish the diagnosis of FHM.
Introduction
Familial hemiplegic migraine (FHM) is a rare subtype of migraine with aura, characterized by attacks of transient hemiplegic aura and an autosomal dominant pattern of inheritance (1). Mutations in three different causative genes have been identified in affected pedigrees: CACNA1A (FHM1), encoding the pore-forming subunit of a neuronal, voltage-gated calcium channel; ATP1A2 (FHM2), encoding an astrocytic sodium-potassium pump; and SCN1A (FHM3), encoding the alpha subunit of a voltage-gated neuronal sodium channel. We describe three families with mutations in the ATP1A2 gene: one large three-generation family with a new mutation segregating completely with the phenotype as well as two other unrelated families with a mutation previously described in a single patient with sporadic HM (SHM) (2).
Patients and methods
Between 2003 and 2012 we observed three families with FHM2 in the Department of Neurology, Klinikum Kassel, Germany. Index patients of each of the three families were admitted to our hospital because of severe FHM attacks. The diagnosis was based on the International Classification of Headache Disorders (1). After obtaining written informed consent, genetic analyses were performed in eight clinically affected patients as well as in four unaffected relatives and in one relative with mental retardation (MR). In our department, patients with a history of migraine associated with hemiplegia and an autosomal dominant pattern of inheritance are typically first tested for mutations in the CACNA1A gene (FHM1), followed, in case of negative results, by screening of ATP1A2.
DNA was extracted from peripheral blood according to standard protocols. Sequence analysis was performed according to a Sanger sequencing protocol published elsewhere. Sequence data were compared to their corresponding reference sequences (CACNA1A: NM_023035.2; ATP1A2: NM_000702.3) as well as in-house controls. Sequence analysis was performed by Mutation Surveyor V3.10 (www.softgenetics.com). Mutation analysis was performed using Alamut software (www.interactive-biosoftware.com).
Informed consent for collecting data and publication was obtained for all of the examined patients. The study has been presented to our institutional review board at the Philipps University of Marburg but did not require approval because of the retrospective nature of the study. Pedigree charts were made with PCD Pedigree Chart Designer, a free software from Center of Genomics and Transcriptomics (http://pedigree.cegat.de).
Results
The pedigrees of all three families (A–C) are shown in Figures 1–3. Information about a total of 47 family members was collected either by personal examination (n = 15) or indirect anamnesis from relatives (n = 32). Figures 1–3 specify how the information was obtained for each family member. Signs and symptoms of the affected family members are presented in Table 1. None of our patients showed ataxia during or between hemiplegic attacks. None of the examined patients showed signs of a persistent cerebral infarction clinically or in their history. Furthermore, magnetic resonance imaging (MRI) was without signs of acute or previous strokes in all three index patients.
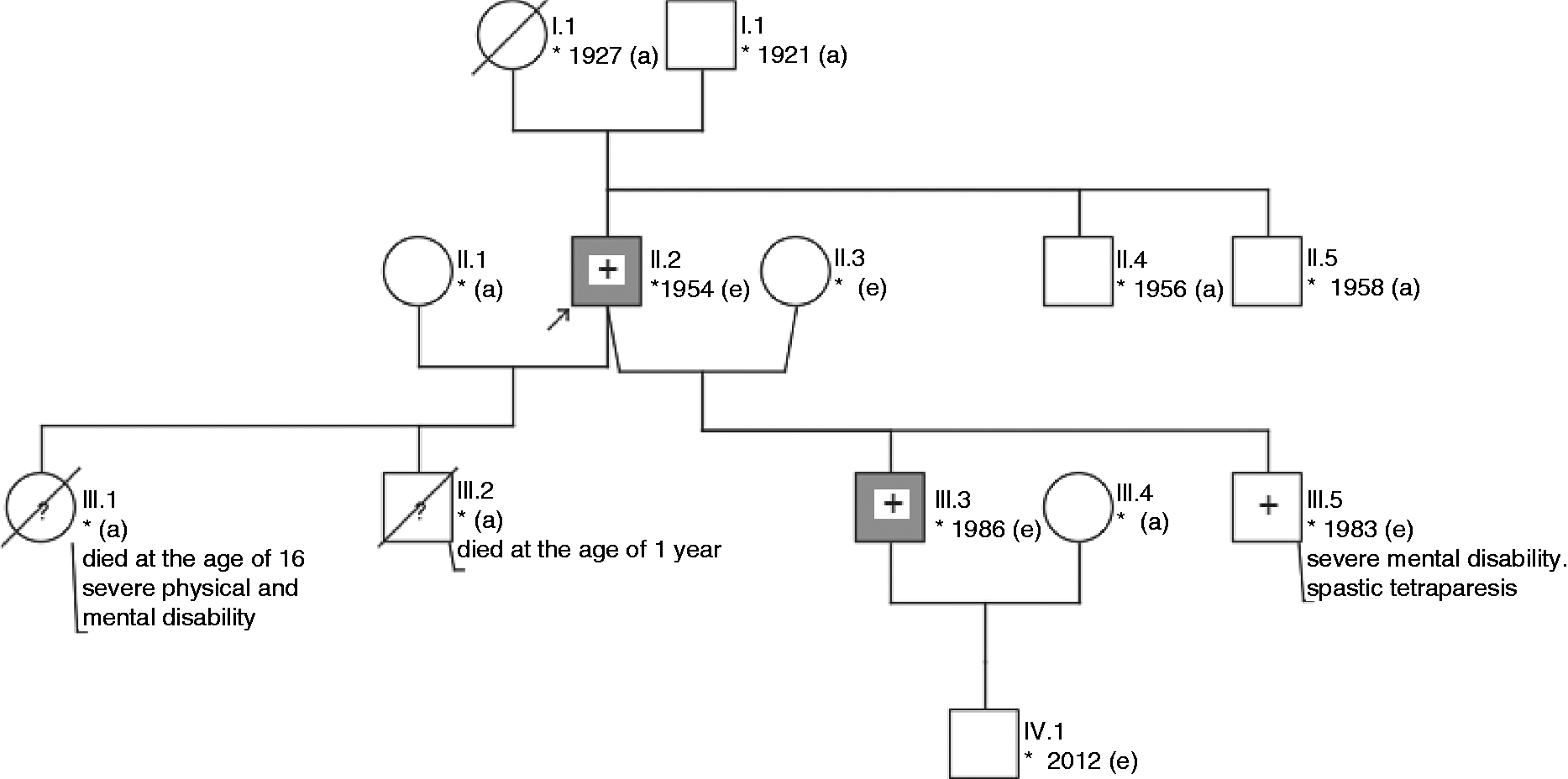
Pedigree of Family A with a novel missense mutation c.659C>T (p.Ser220Leu) in the ATP1A2 gene. Patient IV.1, who had only “occasional headache” but no full-cut FHM phenotype, was also a carrier of the described mutation. II.1 and IV.3, both clinically unaffected, did not show the mutation in the ATP1A2 gene. Individuals reporting migraine are marked in gray; white: healthy; white and ?: history of migraine remains unclear; +: mutation in the ATP1A2 gene; –: no mutation in the ATP1A2 gene; square: male; circle: female; arrow: propositus +; crossed: deceased; asterisk: year of birth; e: personal examination; a: indirect anamnesis of the relatives. Pedigree of Family B with a mutation c.2723G>A, p.Arg908Gln, in the ATP1A2 gene. III.4 and IV.2 were clinically unaffected and did not show the described mutation. IV.6 had a history of migraine attacks since the age of 9. She committed suicide at the age of 32. Individuals reporting migraine are marked in gray; white: healthy; white and ?: history of migraine remains unclear; +: mutation in the ATP1A2 gene; –: no mutation in the ATP1A2 gene; square: male; circle: female; arrow: propositus +; crossed: deceased; asterisk: year of birth; e: personal examination; a: indirect anamnesis of the relatives. Pedigree of Family C. Analysis of the ATP1A2 gene in three family members revealed the mutation c.2723G>A, p.Arg908Gln. II.2 and III.3 were clinically affected, III.5 showed severe mental disability and spastic tetraparesis. Taking the past medical history was difficult. However, the parents denied signs of headache or hemiparesis. Individuals reporting migraine are marked in gray; white: healthy; white and ?: history of migraine remains unclear; +: mutation in the ATP1A2 gene; –: no mutation in the ATP1A2 gene; square: male; circle: female; arrow: propositus +; crossed: deceased; asterisk: year of birth; e: personal examination; a: indirect anamnesis of the relatives. Clinical characteristics of nine patients of three different families (A–C) with mutations in the ATP1A2 gene including a novel mutation in family A. F: female; M: male.


Family A
The index patient (Figure 1, Family A, IV.2) was referred to our emergency department in December 2010 (at this time at the age of 19 years) with extreme migraine-type headache, nausea, vomiting and photophobia. On admission, he showed moderate left-sided hemiparesis without aphasia or visual signs. Body temperature was 38℃ with elevated infection parameters (C-reactive protein 36 mg/l (<5 mg/l); leukocytes 11.800/µl (4000–10,000/µl)). During the first days he showed rude and verbally aggressive behavior. Wearing his night dress, he left the ward twice without previously informing the staff. Cerebrospinal fluid was normal. Repeated electroencephalogram (EEG) recordings showed intermittent delta activity over the right hemisphere during the attack that was no longer present on follow-up EEG after resolution of symptoms. The patient was treated with para-acetylaminophenol and acetylsalicylic acid without relief. Therefore, a therapeutic attempt with intravenous (IV) corticosteroids was initiated. Symptoms resolved completely within six days, and his behavior was friendly and open-minded again. This severe attack was the only one lasting for several days. Migraine with aura was known since the age of 20 with attacks occurring three to four times a year; aura symptoms typically included aphasia lasting for 20 minutes, while hemiparesis was present only rarely. The parents reported that an intelligence quotient (IQ) test that had been given in childhood because of delayed physical development, and language impairment had been below normal.
A detailed family history revealed two further affected family members (Figure 1). The mother (III.2) and grandmother of the index patient (II.2) suffered from recurrent migraine-type headache with associated hemiplegia and/or aphasia. The mother of the propositus (III.2) was a 45-year-old woman who was seen in our outpatient department and was symptom free at the time of examination. She reported migraine-type headache with aura since she was 8 years old. The aura consisted of visual disturbance, aphasia and hemiparesis lasting for about 20 minutes followed by severe headache for 12 hours. Prolonged symptoms for several days, as seen in her son, could not be observed. Since the frequency of headaches was limited to two to three attacks/month she did not currently take any prophylactic medication. The grandmother (II.2) had recurrent episodes of severe and frequent migraine attacks with vomiting but without any signs of aphasia or hemiparesis in her youth. Since her menopause the attacks of headache are rare.
First, a blood sample of the mother was obtained (III.2). No mutation could be detected in the CACNA1A gene; by contrast, an unknown mutation c.659C>T (p.Ser220Leu) in the ATP1A2 gene could be found (Figure 1, Table 1). Segregation analysis was performed and all three family members with a history of migraine showed this mutation (II.2, III.2, IV.2). Furthermore, one brother of the propositus (IV.3) and a brother of the grandmother (II.3) could be examined. Both were clinically unaffected and genetic analysis of the ATP1A2 gene did not show the described mutation in any of them. Finally, the second brother of the propositus (IV.1) was examined. He occasionally complained about holocephalic headache without any aura, especially without hemiparesis once a year, not fulfilling the criteria for migraine without aura. He also showed the mutation c.659C>T in the ATP1A2 gene.
Family B
The propositus, a 45-year-old female (Figure 2, Family B, IV.5), was seen as an outpatient at our institution in 2006, when she complained of an intense throbbing holocephal headache, right-sided numbness of the arm and leg, right-sided facial paresis and nonfluent aphasia. All symptoms completely resolved within half an hour by the time she was seen by a neurologist. A neurological examination and routine blood panel revealed no abnormalities. She was admitted again to our clinic in March 2010 with severe headache with nausea and vomiting accompanied by marked nonfluent aphasia. Clinical examination on admission further revealed only a mild facial paresis and hemihypaesthesia on the right but no motor weakness. Ultrasound of the cerebral arteries was normal on admission but EEG demonstrated delta wave activity over the left hemisphere. Symptoms gradually improved within the next day and resolved completely. Her father (III.1) and her sister (IV.6) also showed a history of migraine with transient hemiparesis and aphasia. The 70-year-old father complained about migraine-type headache since he was 6 years old and also about transient hemiparesis during some of those attacks. A sister of the propositus had a history of migraine in combination with hemiparesis since she was 9 years old. She died from suicide at the age of 32. Blood samples were not available. No further data regarding age of onset or clinical symptoms were available.
Interestingly, sequencing of the CACNA1A gene in three family members (III.1, IV.2, IV.5) revealed the sequence variation c.6800G>A (p.Gly2267Asp), which has not been described yet. However, this change was also found in another 68-year-old family member who did not suffer from any headache at all (III.4). Furthermore, this patient reported that there were no migraine cases in her direct family, neither in her mother (II.4), brother (III.5) nor her two children (IV.7, IV.8). Further genetic analysis was performed on blood samples from the propositus (IV.5) and her father (III.2) that revealed a mutation c.2723G>A (p.Arg908Gln) in the ATP1A2 gene in both of them. A further patient complained about occasional headache once a year but without any aura (IV.2). Diagnostic criteria for migraine without aura were not fulfilled. This patient as well as the patient without any history of headache (III.4) did not show this mutation in the ATP1A2 gene.
Family C
The propositus, a 58-year-old male, (Figure 3, Family C, patient II.2) was admitted to our department twice in 2003 and 2012 because of severe attacks of headache and fever up to 39℃ in combination with hemiparesis and drowsiness. During the first admission, he showed moderate left-sided hemiparesis, whereas the contralateral side was affected during the second episode. During both episodes, symptoms deteriorated over three to four days and the patient showed a hemiplegia in combination with global aphasia and coma. In 2012, he developed complex focal and generalized seizures on day 7 of his hospital stay. Under antiepileptic therapy (benzodiazepines, valproic acid and levetiracetam), no further seizures were observed. Interestingly, cerebrospinal fluid analysis revealed mild pleocytosis (10/µl, reference <5/µl) with normal protein, glucose and lactate levels. EEG revealed transient theta-delta activity over the frontotemporal regions contralateral to the hemiparesis and, notably, epileptic activity in the form of sharp waves in this area. Overall the symptoms persisted for several weeks (three weeks during the first episode and six weeks during the second episode) and resolved completely without neurological signs or symptoms after both attacks. The patient did not show any signs of mental disability or cerebellar ataxia. He reported having recurrent episodes of migraine since he was 10 years old with infrequent aura symptoms in the form of hemiparesis, aphasia and/or visual disturbance lasting only up to 30 minutes before occurrence of headache. The side of hemiparesis was alternating during the attacks.
The patient had four children (Figure 3). Both sons from his first marriage died (III.1, III.2), one at the age of 1 year and the other at the age of 16 years. Causes of death were unknown but the younger son was known to have a mental disability. A 26-year-old son of his second marriage (III.3) also suffered from migraine with visual and hemiplegic aura since the age of 10. The expressions of the symptoms during his attacks were diverse. Since the age of 26 he has had three severe attacks with symptoms lasting over several weeks. Beside those he also has had migraine attacks beginning with an aura followed by headache after the neurological symptoms resolved. The aura always became manifest with a hemiparesis alternating from the left or the right side. He is now on a medication with metoprolol whereby the frequency of the attacks has decreased from once per month to once every three months. The other son had severe MR and delayed physical development since early childhood (III.5). He never learned to walk. During examination in our outpatient department he showed a spastic tetraparesis without intelligible speech. At the age of 10 he developed generalized seizures and is still on valproic acid. Asked about further signs of headache or hemiparesis, the parents denied that he had had any. An array comparative genomic hybridization (CGH) analysis, which was performed in order to find a possible cause for mental disability, did not reveal a microdeletion.
An analysis of the CACNA1A gene did not show any abnormalities in the propositus (II.2). Genetic test of the ATP1A2 gene in the three family members described above (II.2, III.3 and III.5) revealed the mutation c.2723G>A (p.Arg908Gln).
Discussion
We present clinical and genetic data on three families with FHM2. In Family A we identified a novel missense mutation in the ATP1A2 gene (c.659C>T, p.Ser220Leu). In two other families (B and C), unrelated and with different ethnic backgrounds (one from Germany and one from Russia), we detected a mutation in the ATP1A2 gene (c.2723G>A, p.Arg908Gln). Of note, this mutation gene has been previously reported in a single patient with SHM (case 34 in de Vries et al. (2)). Several lines of evidence support the pathogenicity of the identified mutation: As indicated by the multiple sequence alignment, Ser220 is completely conserved among different human and nonhuman members of the Na + /K + -ATPase family. The mutation is located within the first large intracellular loop of the A1A2 protein, which also contains a few other FHM2 mutations (e.g. Arg202Gln, Thr263Met). This loop is part of the so-called A (i.e. actuator) domain of the protein, which is important for dephosphorylating the P domain of the protein using a highly conserved TGES motif of which Ser220 is a part. In line with these arguments, PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) provides a score of 0.987 (i.e. “probably damaging”) for Ser220Leu.
We would like to emphasize that the ATP1A2 variant was found in only the two clinically affected family members but not in clinically unaffected patients, while the CACNA1A variant was found in two affected family members and a further family member without any history of headache, which strongly argues in favor of the pathogenicity of the ATP1A2 variant. More important, the ATP1A2 R908Q was previously functionally evaluated, with clear-cut evidence of a loss-of-function effect (2,3). The two families described here add further evidence for this variant being a causative mutation. Moreover, the PolyPhen2 tool rates Gly2267Asp with a score of only 0.052, corresponding to “benign.” We suspect the mutation in the CACNA1A gene to be a single nucleotide polymorphism (SNP).
Several aspects of our observations deserve attention: First, the index patients of Families A and C had extraordinarily severe attacks with long-lasting hemiplegia and aphasia ranging from 10 to 30 days. In fact, it was these attacks that led to hospital admission. Such severe and prolonged episodes have previously been described in patients with FHM2 (4,5). The bulk of the attacks in these two probands as well as the other affected family members were shorter or even without aura, which again emphasizes the high degree of inter- and intra-individual phenotypic variability in FHM. However, six weeks of transient hemiplegia and aphasia are in clear contrast to the upper limit of 24 hours as suggested in the definition of FHM by the International Classification of Headache Disorders (1). Thus, FHM is not only different from “normal” migraine with aura with regard to symptoms and signs, but also concerning its temporal pattern. In line with a similar proposal by Russell and Ducros (6), this needs to be taken into consideration for the next adaptation of the International Classification of Headache Disorders.
Secondly, three of our ATP1A2 mutation carriers (Family A, II.2 and IV.1 and Family B, IV.5) did not fulfill the complete diagnostic criteria of FHM, which reinforces the previous observation of reduced penetrance of ATP1A2 mutations. At the same time, it emphasizes the critical significance of taking into account the full family history for establishing the diagnosis of FHM.
Finally, our findings expand on the previous observation of cognitive deficits being part of the phenotypic spectrum of FHM. MR has been described in a few patients with FHM 2 (one case (7), two cases (4) and two cases (8)). Likewise, progressive cognitive decline as well MR have been reported in FHM1 (9,10). Here we observed MR in a total of two of our patients. In one of them (Family C, III.5) MR was severe, and array CGH analysis did not reveal a microdeletion (i.e. one of the most prevalent causes of MR), which strengthens the assumption that MR was causally related to the ATP1A2 genotype. Interestingly, this mutation carrier had no history of HM attacks. Another patient (Family A, IV.2) showed delayed physical development and language impairment since childhood, while none of the other family members affected by FHM2 showed MR. Taking together the cases from the literature and our patients, MR can be regarded as part of the spectrum of FHM2. However, the fact that the majority of FHM2 patients (including the patient previously reported with the p.Arg908Gln mutation) do not have cognitive deficits suggests that additional (genetic or environmental) modifying factors are required for the manifestation of the MR phenotype in FHM2.
In conclusion, we describe three new families with FHM2 and one new mutation in ATP1A2. The phenotypic spectrum included not only severe attacks with deficits lasting up to six weeks and epileptic seizures but also cognitive deficits in some patients. Further, FHM2 may also present without hemiplegic aura in some patients.
Clinical implications
The missense variant in the ATP1A2 gene (c.659C>T, p.Ser220Leu) is a novel mutation in familial hemiplegic migraine type II (FHM2). Clinical symptoms, such as hemiplegia or aphasia, may last for several weeks in some patients. Patients with FHM2 may also present without hemiplegia. Therefore, the full family history has to be taken in account to establish the diagnosis of FHM.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
None declared.