
Abstract
Purpose of review
Migraine is a prevalent neurovascular brain disorder with a strong genetic component, and different methodological approaches have been implemented to identify the genes involved. This review focuses on pearls and pitfalls of these approaches and genetic findings in migraine.
Summary
Common forms of migraine (i.e. migraine with and without aura) are thought to have a polygenic make-up, whereas rare familial hemiplegic migraine (FHM) presents with a monogenic pattern of inheritance. Until a few years ago only studies in FHM yielded causal genes, which were identified by a classical linkage analysis approach. Functional analyses of FHM gene mutations in cellular and transgenic animal models suggest abnormal glutamatergic neurotransmission as a possible key disease mechanism. Recently, a number of genes were discovered for the common forms of migraine using a genome-wide association (GWA) approach, which sheds first light on the pathophysiological mechanisms involved.
Conclusions
Novel technological strategies such as next-generation sequencing, which can be implemented in future genetic migraine research, may aid the identification of novel FHM genes and promote the search for the missing heritability of common migraine.
Introduction: Migraine as a genetic disease
Migraine is a common neurovascular brain disorder that is characterized by attacks of severe, unilateral, pulsatile headache that is often accompanied by nausea, vomiting, photo- and/or phonophobia (1). Women are three times more often affected than men (2). At least 12% of the general population suffers from recurrent migraine attacks (3). The presence or absence of an aura can precede the headache phase in one-third of patients, and distinguishes between migraine with aura (MA) and migraine without aura (MO) (1). The aura consists of transient mainly visual, sensory and speech-related symptoms, and is likely caused by cortical spreading depression (CSD) events, which are slowly propagating cortical waves of neuronal and glial depolarization (4,5). The headaches are brought about by an activation of the trigeminovascular system that involves abnormal processing of signals from innervated blood vessels in the meninges to brainstem centers. Studies in experimental animals have indicated that CSD events may activate the relevant brainstem centers and thereby would link aura and pain mechanisms (6–8), but such proof is lacking in humans.
Migraine has a strong genetic component. Population-based family studies showed that the familial risk of migraine is increased (9,10). A contribution of genetic factors in migraine was also apparent from twin studies that showed a concordance twice as high in monozygotic versus dizygotic twins (11,12). In fact, a large Dutch twin study revealed that genetic and environmental factors had an almost equally large contribution (13). This review highlights some of the recent advances and controversies in the search for migraine genes. Special attention is paid to the different methodological approaches used in genetic research in the (recent) past (linkage with markers, candidate gene association studies), the present (genome-wide association studies (GWAS)), and the future (next-generation sequencing (NGS)).
Pearls in genetics
Gene identification by classical linkage analysis in monogenic familial hemiplegic migraine (FHM)
A linkage approach, traditionally, has been most successful for the identification of causal genes in monogenic disorders. With linkage analysis the segregation of hundreds to several thousand genetic markers (earlier multi-allelic markers and more recently single-nucleotide polymorphisms (SNPs)) that are evenly distributed over the genome are compared with the co-occurrence of a disease trait in a family-based design. The fit is calculated as a logarithm (base 10) of odds (LOD) score that provides statistical proof that a trait locus resides at a given chromosomal location. Subsequent sequencing of candidate genes in the linked chromosomal region is needed to identify the causal gene mutation in patients that should not be present in control subjects. In migraine genetics the linkage approach was successfully applied for FHM, a rare autosomal dominant subtype of MA that is characterized by a transient hemiparesis during the aura phase of the attack, which led to the identification of three genes.
FHM genes CACNA1A (FHM1) (14)), ATP1A2 (FHM2) (15) and SCN1A (FHM3) (16) all encode proteins that affect ion transport in the brain. Their gene products are subunits of certain voltage-gated calcium channels, sodium-potassium ATPases, or voltage-gated sodium channels, respectively, and analysis of gene mutations identified a major role for glutamatergic neurotransmission in migraine (Figure 1) (17). The identification of FHM genes has had an enormous impact on diagnosing patients as the presence of a gene mutation, with its large effect size, confirms the clinical diagnosis. Genotype-phenotype correlation studies revealed that mutation carriers can suffer from a wide variety of associated symptoms, including cerebellar ataxia, seizures, mental retardation, mild head trauma-induced edema that can be fatal (e.g. in case of the FHM1 S218L missense mutation (18)), or even “elicited repetitive daily blindness” that can occur apart from FHM attacks in some carriers of FHM3 mutations Q1489H and F1499L (19,20). Notably, certain mutations in FHM genes do not cause FHM, but seemingly unrelated brain disorders, such as episodic ataxia type 2 or spinocerebellar ataxia type 6 (in the case of CACNA1A) (14,21) or severe childhood epilepsies, such as severe myoclonic epilepsy of infancy and generalized epilepsy with febrile seizures plus (in the case of SCN1A) (22), which provides additional opportunities to investigate how FHM gene mutations affect brain function in a disease-specific manner.
Genes and pathways involved in familial hemiplegic migraine (FHM) and common migraine. FHM genes CACNA1A (FHM1), ATP1A2 (FHM2) and SCN1A (FHM3) are involved in the regulation of (glutamatergic) neurotransmission. Common migraine genes MTDH, LRP1 and MEF2D that are indicated by genome-wide association studies (GWAS) hits seem to be involved in this pathway as well. Additional common migraine genes from the GWAS indicate neuron and synapse development (PHACTR1, ASTN2, PRDM16, TGFBR2), brain vasculature function (TGFBR2, PHACTR1) and pain-sensing (TRPM8) as additional migraine-relevant pathways.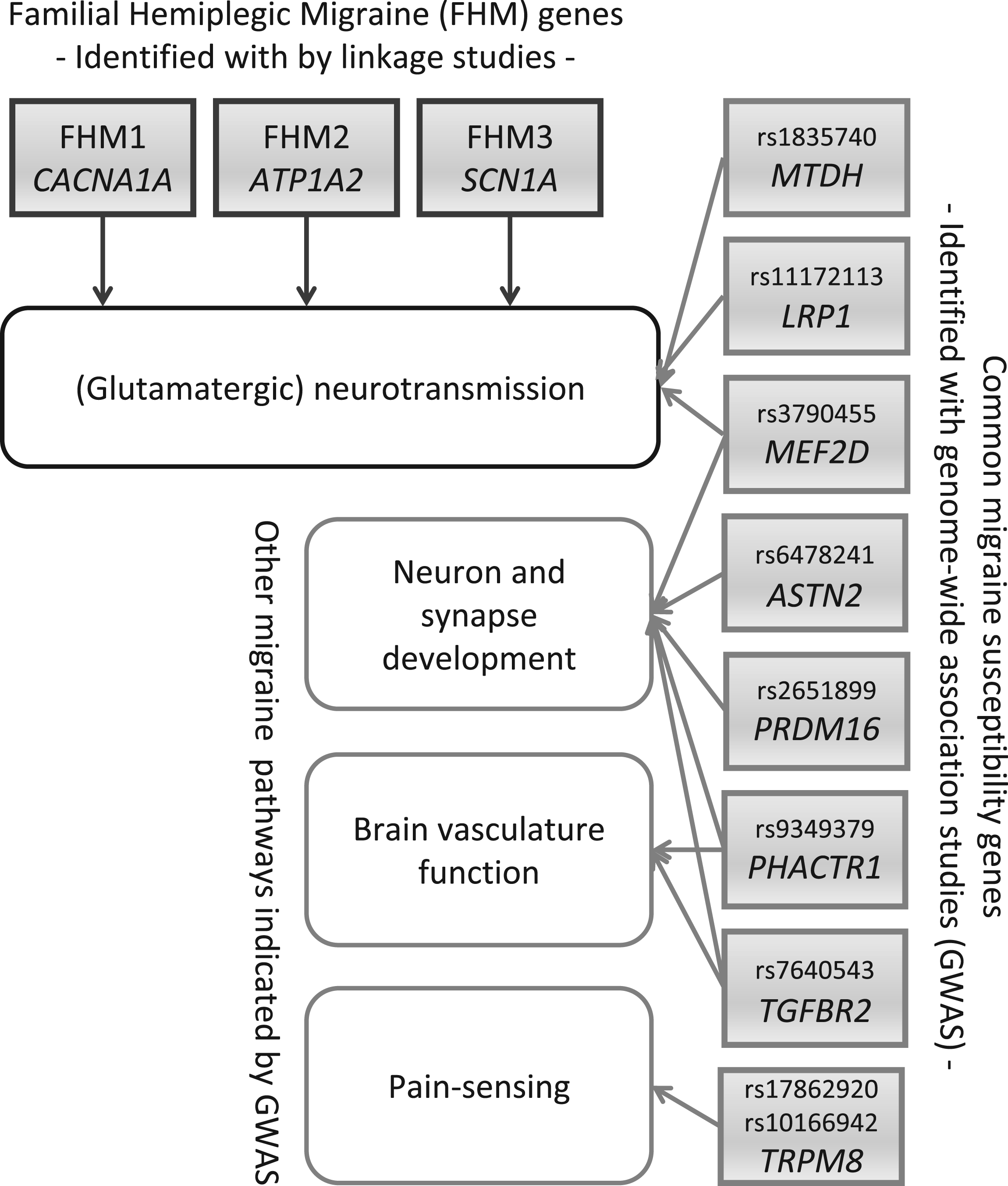
If hemiplegic migraine occurs not in a family but in an isolated case, it is called sporadic hemiplegic migraine (SHM). Apart from the absence of an affected close relative, the diagnostic criteria and attacks are identical to those in FHM (23). Most screens of FHM genes in SHM patients revealed mutations, predominantly in ATP1A2, in only a small proportion of patients (24–27). Riant and colleagues (28) studied a group of 25 SHM patients with an age of onset before 16 years, of whom 18 patients had additional symptoms such as epilepsy, learning difficulties, cerebellar ataxia, and/or coma. Perhaps surprisingly, in no fewer than 23 patients, mutations in CACNA1A and ATP1A2 were identified. Three-quarters of the mutations had occurred de novo and mutation carriers thus represent the first patients of new FHM families. A recently identified de novo G715R ATP1A2 mutation was found in a 6-year-old SHM patient with prolonged hemiparesis, which resolved after four weeks and required rehabilitation (29); this further supports the observation that particularly de novo mutations are present in severely affected patients. The question remains what is causing SHM in the patients who do not carry an FHM gene mutation, which is a rather large proportion of patients in most studies. Possibilities are that either other FHM genes may cause hemiplegic migraine in those patients or that SHM (especially when the phenotype is not severe) is due to a combination of multiple low-risk genetic variants, similar to what is predicted to occur in common migraine. Support for the latter hypothesis comes from the observation that MA is frequent in families of SHM patients (30). Also, it would fit a view of migraine being a spectrum of disorders. It certainly stresses the importance of taking a reliable family history in patients with suspected hemiplegic migraine and of follow-up of these patients. This is also exemplified by a recent clinical follow-up study of 19 Dutch SHM patients, which showed that in a proportion of SHM patients the diagnosis changed over time to FHM (31).
Genetic studies that tried to find evidence for involvement of FHM genes in common forms of migraine are essentially negative, including the most systematic and largest study that screened several thousand DNA polymorphisms in more than 150 ion transporter genes in close to a thousand Finnish MA patients (and several thousand additional migraine patients in the replication phase of initial slightly positive association finding) (32). Of note, a similar-sized study in epilepsy also did not find genetic support for the involvement of ion transporter genes (33). This may seem paradoxical, also in light of the efficacy of anti-epileptic drugs in epileptic and migraine patients, which act on neurotransmitter and ion channel pathways. A possible explanation might be that disease risk in these disorders is conferred by other — perhaps regulatory — genes that control neurotransmitter and ion pathways in a more subtle manner than the ion transporters themselves.
Functional characterization of FHM mutations in cellular and transgenic mouse models
The large effect sizes of gene mutations in monogenic FHM make an investigation of their functional consequences in cellular and transgenic animal models feasible. Most cellular studies showed that FHM1 mutations exert gain-of-function effects by shifting neuronal CaV2.1 channels’ voltage-dependence toward more negative membrane potentials while enhancing channel open probability (34), although loss-of-function effects have been reported as well (35,36). Loss of glial Na+/K+ ATPase function seems the most likely mechanism for FHM2 mutations (37). Most FHM3 mutations seem to exert loss-of-function effects of NaV1.1 sodium channels that seem to primarily affect inhibitory neurons, but gain-of-function effects have been proposed for FHM3 mutation L263V that is associated with a combined seizure FHM phenotype in the majority of mutation carriers (38–40). Taken together, these cellular studies of FHM mutations predict increased neurotransmitter and potassium ion levels at the synaptic cleft, especially after high-intensity neuronal firing, which would facilitate cortical spreading depression (CSD) (41), and thereby could explain the migraine aura.
In vivo consequences of FHM mutations at the organism level can be investigated with the use of so-called knock-in (KI) mouse models by introducing human pathogenic mutations in the endogenous gene using gene-targeting technology. For FHM1, two KI models have been generated that harbor gain-of-function missense mutations R192Q or S218L (42,43). In line with the clinical phenotype in mutation carriers, only S218L mice exhibit the complex phenotype of cerebellar ataxia, susceptibility to seizures and delayed cerebral edema after minor head trauma (43). Mice with the milder R192Q mutation show no overt phenotype (42), although signs of abnormal facial expression that may reflect a pain phenotype, which was prevented by administrating a serotonergic migraine drug, have been reported (44). At the neurobiological level, mice of both mutant strains revealed increased neuronal calcium influx and (cortical) neurotransmitter release, as well as an increased susceptibility to CSD (42,43,45,46). These features were all more prominent in the severer S218L mutant. In line with the female preponderance in migraineurs, female mutant mice are more susceptible to CSD than male mice (45). Recently, increased susceptibility to CSD was also reported in heterozygous KI mice that harbor the loss-of-function FHM2 mutation W887R (47). The experiments cannot be conducted in (adult) homozygous FHM2 mutant mice as the mice die directly after birth, identical to the fate of homozygous Atp1a2 knockout mice (48).
GWAS revealed first susceptibility genes in common migraine
During the last few years, GWAS have become the most used approach to identify genes that confer susceptibility to complex disorders. In a GWAS, hundreds of thousands of SNPs that are distributed over the genome are tested in a hypothesis-free manner for association with a disease trait. For each SNP, the allele frequencies are compared between cases and controls. Significant differences in allele frequency either pinpoint the SNP itself as a genetic susceptibility factor or provide statistical evidence that a causal gene variant is in close vicinity. The latter implies that the tested SNP and the causal variant do not segregate independently; in genetic terms they are in linkage disequilibrium. To sufficiently correct for multiple testing (i.e. for the hundreds of thousands of tests), only SNP-trait associations with p values below 5.0 × 10−8 are considered genome-wide significant. As a direct consequence, GWA studies require testing of several or many thousand cases and controls to have sufficient power. Importantly, initial positive association findings need to be confirmed in independent case and control cohorts before meaningful conclusions about the associations can be drawn.
Migraine loci and genes identified by genome-wide association studies.
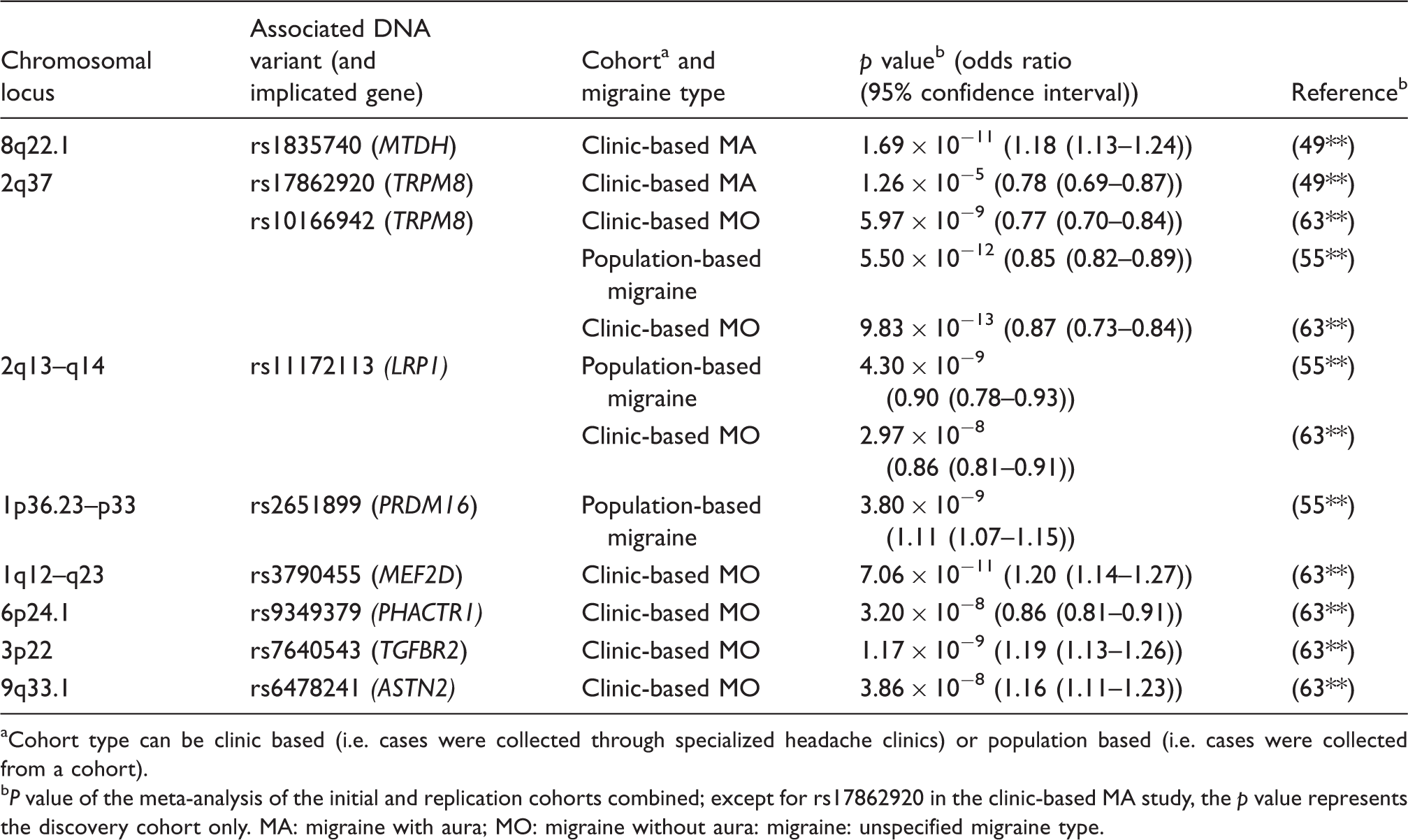
Cohort type can be clinic based (i.e. cases were collected through specialized headache clinics) or population based (i.e. cases were collected from a cohort).
P value of the meta-analysis of the initial and replication cohorts combined; except for rs17862920 in the clinic-based MA study, the p value represents the discovery cohort only. MA: migraine with aura; MO: migraine without aura: migraine: unspecified migraine type.
The second major GWAS was conducted as part of the Women’s Genome Health Study and involved 5122 migraine cases and more than 18,000 controls from the general population (55**). After including the replication data, three loci became genome-wide significant, suggesting TRPM8, LRP1 and PRDM16 as migraine susceptibility genes. The association findings were strongest for TRPM8. TRPM8 is a calcium ion channel protein that is located on peripheral afferents of sensory neurons and a subset of trigeminal ganglion neurons and is a sensor for cold and cold-induced burning pain (56,57). TRPM8 is a target in animal models of neuropathic pain (58). LRP1 encodes a lipoprotein receptor that seems to modulate neuronal glutamate signaling by astrocytic cycling of tissue-type plasminogen activator (59). A possible role for PRDM16 in migraine pathophysiology is less clear as the gene is implicated in brown fat development (60) although recent studies indicated a possible role in neuronal development as well (61,62). The third GWA study was conducted in clinic-based MO patients (63**). The study included 2326 patients and almost 5000 controls, and identified four novel migraine susceptibility genes: MEF2D, TGFBR2, PHACTR1 and ASTN2, of which the latter two had less convincing p values. In addition, this study confirmed a role for TRPM8 and LRP1 in migraine pathophysiology. At this moment, it is speculative how the novel genes may affect migraine pathophysiology. Of note, MEF2D encodes a transcription factor that controls synapse development (64). Furthermore, one of the target genes of MEF2D encodes pituitary adenylate cyclase-activating polypeptide-38 (PACAP-38), which may modulate excitatory synaptic transmission and can trigger migraine-like attacks in persons with MO (65). TGFBR2 encodes a serine-threonine kinase that is involved in cell proliferation and differentiation, as well as extracellular matrix production. A vascular role of TGFBR2 in migraine can be hypothesized from the observation that missense mutation p.Arg460His in TGFBR2 causes familial aortic dissection as well as migrainous headaches in 11 of 14 mutation carriers in a large multigenerational family (66). Although still highly speculative, PHACTR1, which is a member the PHACTR/scapinin family, might play a role in synaptic activity and synapse morphology or in endothelial cell functioning (67).
As the odds ratios observed with GWA studies in complex diseases generally are very small, ranging from 1.05 to 1.25, the identified genetic variants have no direct clinical relevance for patient care. The same is true for migraine. This is not surprising since variants identified by GWA studies are present in a considerable proportion of healthy subjects. The variants occurred early in human evolution and it is therefore unlikely that they would have survived during evolution if they would have affected disease susceptibility in a dramatic manner. As many disease variants are located outside genes, for many of them it is not even immediately evident which biological pathways may be affected in patients. Still, hundreds to thousands of genes that have surfaced from the hundreds of GWA studies for a plethora of complex disorders provided first important insight in possible disease mechanisms. The low effect size is a complicating factor and considerably hampers straightforward functional analysis of disease-associated gene variants. Multiple ways have been proposed to bypass this downside of GWAS findings. For instance, one could perform biological pathway analyses of groups of related genes in specific pathways, guided by GWAS findings (68). Alternatively, one could investigate the function of the identified genes themselves for their consequence on disease-relevant pathways by modulating their expression (i.e. overexpression or knockdown) in various cellular and/or animal models (e.g. zebrafish or mouse) (69). Surely, development of more sophisticated models, which may include differentiated human-induced pluripotent stem cells as has been proposed for schizophrenia (70), seems needed for a meaningful functional evaluation of GWAS gene hits.
Pitfalls
Classical linkage approach in common forms of migraine
Whereas classical linkage analysis in monogenic FHM has led to the identification of several migraine genes, similar studies investigating common forms of migraine were not so successful. Although many chromosomal regions were identified that seem to harbor migraine susceptibility genes, no migraine genes were discovered (71). The most likely reason is that the genetic make-up in migraineurs with common migraine types is more complex with many genetic factors determining disease susceptibility together with environmental factors. Migraine susceptibility loci that reside on chromosomes 4q21–q24 (72,73) and 10q22–q23 (74,75**,76) seem most promising, also because these loci were found in more than one study. Still, the area of classical linkage approach led to potentially important novel ideas for phenotyping patients for genetic studies by challenging the classically used end-diagnoses of MA and MO as defined by the International Criteria of Headache Disorders (ICHD-II) (1). Using “latent class analysis” (LCA) that involves complex statistical empirical clustering based on factor analysis combining information on several migraine symptoms (77**) or analyzing individual migraine sub-traits in “trait component analysis (TCA)” (75**) may yield better results in future genetic migraine studies. Genetic studies of other complex disorders (e.g. schizophrenia and attention-deficit/hyperactivity disorder (ADHD)) have already indicated that individual sub-traits may have a higher heritability than the end-diagnosis itself (78,79).
Candidate-gene association studies in migraine
Before the GWAS era, a frequently used alternative to family-based classical linkage studies has been the investigation of one (or sometimes a few) DNA variants in candidate genes that had emerged from pre-existing knowledge. Many genes of the dopaminergic and serotonergic systems, hormone receptors and genes in inflammatory pathways were investigated (for a review, see Proudfoot et al. (57)). Although candidate gene-based association studies, in theory, present a powerful tool, they did not yield robust genetic associations. Most studies have one or more methodological issues with respect to small sample size, selection of cases and controls, insufficient correction for multiple testing, and/or incapability to replicate findings in independent populations. The best replicated genetic association is the C677T polymorphism in the 5,10-methylenetetrahydrofolate reductase (MTHFR) gene as can be concluded also from two meta-analyses that showed an association of the T-allele with MA, but not MO (80,81). However, neither several large well-designed studies (82,83) nor GWAS data found support for a genetic association. Regardless of lack of genetic evidence, a role for MTHFR, which codes for an enzyme of the homocysteine and folate metabolism (84), is appealing as high homocysteine levels (like seen in carriers of the T-allele) may induce vascular endothelial dysfunction and thereby increase migraine risk (85).
Caution is needed when sequencing candidate genes
Where hypothesis-driven candidate gene association studies in common disorders have led to genetic associations that could not be replicated—and most now are considered false-positive findings—the same may apply to candidate gene-sequencing studies in monogenic disorders if there is no sufficient evidence from replication efforts and if one is not cautious enough about the interpretation of results.
For example, common diseases (or symptoms of these diseases) such as migraine may co-occur just by chance with rare disorders. This fact may have confounded some of the disease implications of a homozygous S982NfsX4-truncating SLC4A4 mutation that resulted in a non-functional sodium bicarbonate co-transporter NBCe1 protein and was identified in two sisters with proximal renal tubular acidosis (pRTA) and ocular abnormalities (86). Whereas mutations in SLC4A4 are a well-known cause of proximal renal tubular acidosis (87), because both sisters also had hemiplegia and migraine attacks, the SLC4A4 gene was also presented as a possible recessive hemiplegic migraine gene. Two of six family members who were heterozygous for the SLC4A4 mutation had migraine (i.e. one had MA, the other had MO), which is not unexpected given the very high frequency of migraine in the general population. Although interesting as an isolated case, the finding may just be a spurious association in spite of the interesting hypothesis that NBCe1 dysfunction may disturb synaptic pH in astrocytes and thereby might affect migraine pathophysiology. The fact that four other homozygous SLC4A4 mutations, in additional to pRTA, cause different migraine phenotypes (i.e. hemiplegic migraine with episodic ataxia, MA, or MO (twice)), seems to add doubt that homozygous SLC4A4 mutations specifically cause hemiplegic migraine, and cannot be considered sufficient replication. It is more likely that (hemiplegic) migraine in these patients is caused by a yet uncovered gene defect.
Although direct sequencing of candidate genes, theoretically, is an appealing genetic approach to identify causal disease genes, one would predict that this is successful only in rare monogenic disorders, which are caused by single gene mutations with a high effect size. Recent large-scale sequencing studies (i.e. NGS of all coding exons in the genome) indicated that each of us, on average, carries a handful of novel nonsense, non-functional variants, which implies that the majority of these variants may be well tolerated (90). According to their publication, Lafrenière and colleagues selected the KCNK18 gene, which encodes the TRESK protein, based on the fact that this member of the two-pore domain family of potassium channels is involved in control of neuronal excitability, as a candidate gene for a multigenerational MA family (88). The F139WfsX24-truncating mutation that was identified results in a functionally inactive TRESK potassium channel protein and seems to fully explain migraine in this single MA family. No further causal evidence came from testing more than 600 additional migraine families. Is this sufficient proof for KCNK18 as a migraine gene? Perhaps not, as the same authors showed that the C110R TRESK missense mutation, which also results in a non-functional copy of the KCNK18 gene, was identified in migraineurs as well as control individuals (89). A logical conclusion seems that non-functional TRESK copies are tolerated and do not lead to disease. KCNK18 mutations may still turn out genetic modifiers of a migraine phenotype, but it is not logical that it explains the clear-cut autosomal dominant inheritance in their original publication. The identification of the TRESK F139WfsX24 mutation seems to originate from a larger study in which 110 unrelated migraine patients were sequenced for 150 ion transporter genes (91), which may have considerably increased the chance of finding a non-functional mutation such as F139WfsX24 that does not have to be disease causing. The more likely scenario is that MA in the presented family is caused by a different gene mutation, perhaps in relative close proximity on chromosome 10q25. Regardless of the genetic doubt about causality, TRESK remains an interesting possible migraine target already because of its role in neuronal excitability (92).
Open questions for the future
Gene identification with NGS
The identification of three FHM genes did not end the gene hunt for this monogenic migraine type as there are many hemiplegic migraine patients in whom no causal gene mutation has been identified. Although hemiplegic migraine is a clinical diagnosis (sometimes compromised when confused with epilepsy or basilar migraine), knowledge of the disease-causing mutation can reassure clinicians and patients of that diagnosis. Moreover, it provides the direct possibility for genetic testing in additional family members. Finally, given the clinical variation observed with certain hemiplegic gene mutations, genetic testing provides information on possible severe clinical outcomes, such as a mild head-trauma-induced lethality (in the case of S218L in the CaV2.1 channel gene CACNA1A (93)) that in many cases would otherwise remain unknown. Moreover, knowledge of the neurobiological mechanisms of known and to-be-identified FHM genes will add further insight to the pathology of FHM, and likely also other migraine types. Current genetic knowledge seems to indicate that monogenic FHM and complex common migraine are caused by different gene variants/mutations with different effect sizes (Figure 2) and may share (at least in part) similar disease mechanisms, such as glutamatergic neurotransmission.
Different categories of genetic variation exist based on the frequency and effect size of a genetic variant. For rare variants with high effect sizes, until recently the classical linkage approach (combined with Sanger sequencing to identify the causal mutation) has been the method of choice for disease gene identification. This led to the identification of CACNA1A, ATP1A2 and SCN1A genes in familial hemiplegic migraine (FHM). Nowadays, next-generation sequencing (i.e. exome and whole-genome sequencing) has replaced the classical linkage approach. Only a few examples (e.g. complement factor H in age-related macular degeneration (102)) are known of genetic factors with a high effect size and allele frequency. Common variants with a low effect size are typically identified with genome-wide association (GWA) studies. Examples of associated single-nucleotide polymorphisms (SNPs) in migraine are shown. Rare variants with a low effect size are currently out of reach; extensive whole-genome sequencing is required for identification of these variants. Discovery of low-frequency variants with medium-size effects might be identified in the next few years using next-generation (re-)sequencing. Adapted from Manolio et al. (99**).
Gene identification capabilities have advanced dramatically in the last couple of years, as truly large-scale sequencing has become feasible by technical advances known as NGS. Whereas until recently genes were individually scrutinized for mutations with low-throughput Sanger dideoxy sequencing, it is now possible to cost-effectively sequence all coding regions of proteins (i.e. exome sequencing) in a single experiment. In the very near future it will become (financially) feasible to sequence the entire genome (i.e. whole-genome sequencing) of multiple patients for research purposes (94**). These novel possibilities will affect foremost monogenic disorders, as the inheritance pattern in these disorders will assist in selecting the most likely causal gene mutation (95**). Importantly, NGS will allow gene identification in FHM families that were too small for classical linkage analysis. It will be challenging though to uncover causal mutations from the vast amount of DNA variants that exome or genome sequencing produces. Sophisticated procedures for data pooling, bioinformatic filtering and variant prioritization methods will be essential to optimally harvest from this novel technology. Until now, most progress has been made with recessive disorders or sporadic disorders in which the mutation presents as de novo (i.e. is present in the patient but absent in unaffected parents). A recent successful example is sporadic alternating hemiplegia of childhood (AHC), which is a rare, severe neurodevelopmental syndrome characterized by recurrent hemiplegic episodes and distinct neurologic manifestations (96). Some 70% of AHC patients were shown to have a heterozygous de novo missense mutation in the ATP1A3 gene (97**,98**) that belongs to the same family as the FHM2 ATP1A2 gene. The real challenge for the future will be to successfully apply NGS to complex disorders and identify causal gene variants with a moderate effect size (i.e. larger than those of GWA studies, but smaller than those in monogenic diseases). For these genetically complex, polygenic disorders, it is far from clear whether analyzing the segregation of the large number of possible causal variants in family-based designs will be of sufficient help to pinpoint causal disease variants.
Recommendation for future studies
The methodology of GWA has been adopted widely in the genetic arena and has led to the identification of thousands of loci (and genes) for a wide variety of common diseases, including migraine. Hypothesis-free GWA studies essentially made candidate gene association studies redundant and definitely raised the bar for them as no genetic association study can do without an independent confirmation in other samples in the same study. Despite the obvious strength of GWA in identifying disease genes, the methodology, in some respects, did not live up to its expectation. Mainly because predicting individual genetic risk of disease from GWAS results is limited. In theory, one should be able to identify individuals with multiple-risk alleles who are at higher risk, but these calculations are not meaningful when only a fraction of the disease genes is known. If one calculates the sum of the effects of all loci discovered for a trait to obtain the combined genetic influence, at best one explains only ∼20% of the heritability of the trait. This implies that ∼80% of the heritability is still missing and not detected by the GWAS approaches used to date (99**). This raises the serious question of where the missing heritability is hidden, and how this can be found. There are several possibilities. It may be that many of the common causal alleles simply have very small effect sizes that can only be detected in a GWAS with much larger sample sizes of cases and controls (i.e. with tens of thousands of individuals each). Also, causal variants may be rare and therefore not covered on commercial SNP chips used for GWA studies. These rare alleles are thought to have a larger effect size and can hopefully be detected by future NGS studies. Defining intermediate and/or functional phenotypes, such as an altered response to a migraine trigger-like nitric oxide donor (100), may be particularly useful for evaluating the genetic contribution of these rarer variants with potentially greater functional effects. Some of the heritability may also be explained by rare copy number variations as seems the case for neuropsychiatric disorders, such as schizophrenia (101**). Finally, one has to take into account mechanisms such as epistasis, gene-environment interactions, and epigenetics. Epistasis can best be explained as variant-variant interactions in which one allele positively or negatively influences another allele, meaning that certain single SNPs with no or low marginal effects may have larger effects only in combination with another SNP (or SNPs). The potential of epistasis has not yet been investigated in migraine, also because the analysis of epistatic interactions suffers from a substantially increased multiple testing burden. Analyses of gene-environment interactions also have been understudied in GWAS, primarily because of lack of data on environmental exposures, which would require large prospective cohorts. Finally, epigenetic modifications, i.e. modifications to the genome other than changes in the DNA sequence, such as DNA methylation and histone modifications, may affect disease susceptibility by influencing gene expression, e.g. after exposure to environmental triggers. An important lesson for the future seems that one would need all of the above-mentioned approaches to further unravel the genetic background of common diseases, such as migraine. Notably, advancing the understanding of the genetic background of diseases that are co-morbid with migraine, e.g. epilepsy and depression, may become instrumental, not only for the understanding of the co-morbid relation, but perhaps also of migraine pathophysiology itself. Undoubtedly, the coming years will be very exciting as the DNA technologies that have been developed in the last few years, e.g. GWAS, exome sequencing and whole-genome sequencing, will be exploited to the full to uncover migraine genes and mechanisms.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
None declared.