
Abstract
Background: Serotonin has an important role in vascular resistance and blood pressure control, and a functional serotonin transporter polymorphism has been associated with migraine. Disturbances in serotonin metabolism have been associated with autism, depression, and myoclonus related conditions, but serotonin has far more functions in the body. Familial hemiplegic migraine is a rare autosomal dominant subtype of migraine with aura in which attacks are associated with hemiparesis.
Cases: We present two siblings with hemiplegic migraine, depression, progressive spastic paraparesis, myelopathy, and spinal cord atrophy. One of the sisters presented with prolonged coma after a migraine episode. Both sisters were found to have low cerebrospinal fluid serotonin metabolite (5-hydroxyindoleacetic acid), low platelet serotonin levels, and diminished serotonin transport capacity. Their clinical symptoms improved on 5-hydroxytryptophan replacement therapy. Mutational analysis of the CACNA1A and ATP1A2 genes was negative.
Conclusion: This is the first time that systemic serotonin deficiency has been described in familial hemiplegic migraine. We hypothesize that the deficiency of serotonin transport may be part of a complex cellular membrane trafficking dysfunction involving not only the serotonin transporter but also other transporters and ion channels.
Introduction
Serotonin (5-hydroxytryptamine or 5-HT) is a monoamine neurotransmitter synthesized in neurons of midbrain Raphe nuclei in the central nervous system and in enterochromaffin cells in the gut. The rate-limiting step in serotonin synthesis is catalyzed by two genetically distinct isoforms of tryptophan hydroxylase: TPH2 is preferentially expressed in the brain, while TPH1 is expressed peripherally and in the pineal gland (1). Serotonin is stored intracellularly in vesicles and granules. Re-uptake of neuronal serotonin across presynaptic plasma membrane is mediated by the serotonin transporter SERT, a member of the SLC6 sodium dependent solute transporter family encoded by the SLC6A4 gene. SERT is highly expressed in platelets that do not synthesize serotonin but store serotonin produced by the enterochromaffin cells.
Disturbances in serotonin metabolism have been associated with autism, depression, and myoclonus related conditions (2,3).
We describe two sisters with familial hemiplegic migraine and neurodegeneration with biochemical findings of serotonin deficiency in the cerebrospinal fluid (CSF) and platelets and a diminished serotonin transporter capacity.
Case reports
Case 1
Case 1 is the third child of non-consanguineous parents. Pregnancy was unremarkable and early development was completely normal.
She started having hemiplegic migraine at age 11 years, initially occurring every 3–8 weeks, lasting 4–48 hours, presenting with right or left-sided numbness and paralysis, no visual disturbances, but slurred speech, associated with vomiting, headache, and confusion, followed by weakness lasting up to 7 days, and then complete recovery. The frequency of her migraine increased slowly with age up to twice a month, and at the age of 16 she had one severe episode requiring prolonged hospitalization, followed by prolonged recovery in a rehabilitation unit. Between 12 and 20 years she had developed progressive spastic paraparesis; sensory loss in stocking distribution, involving sensation of touch, pinprick, temperature, and vibration; urinary and bowel incontinence; bladder instability, diarrhea predominant irritable bowel syndrome; sleep problems; depressed mood; and anxiety. She needed to use a wheelchair for most of the time by the age of 17. At the ages of 15 and 18 she had a benign ovarian tumor and multiple precancerous breast tumors removed. Recently, at the age of 21, she had a severe episode of hemiplegic migraine followed by a 2 month period in deep coma requiring admission to an intensive care unit. Currently she is still recovering and has severe dysarthria and lower leg weakness. This episode happened after 2 months without any migraine and was not preceded by any change of treatment or other precipitating factors, such as head trauma.
Investigations ruled out infectious, inflammatory, or thrombotic processes. Various metabolic investigations gave normal results, including amino and organic acids, creatine, ammonia, lactate, folate, and vitamin B12. Acetylcholine receptor antibodies were negative, and creatine kinase levels were normal. EMG and nerve conduction studies were repeated twice and were normal, with no signs of myopathy or peripheral neuropathy. Muscle biopsy showed mild type 1 fiber predominance, normal mitochondrial histochemistry and structure and normal activities of mitochondrial respiratory chain enzymes. Molecular investigations for myotonic dystrophy (DMPK) and the migraine-related genes CACNA1A and ATP1A2 were negative. Mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS) mutations, with which migraine has been described as an associated feature, were also tested and were negative.
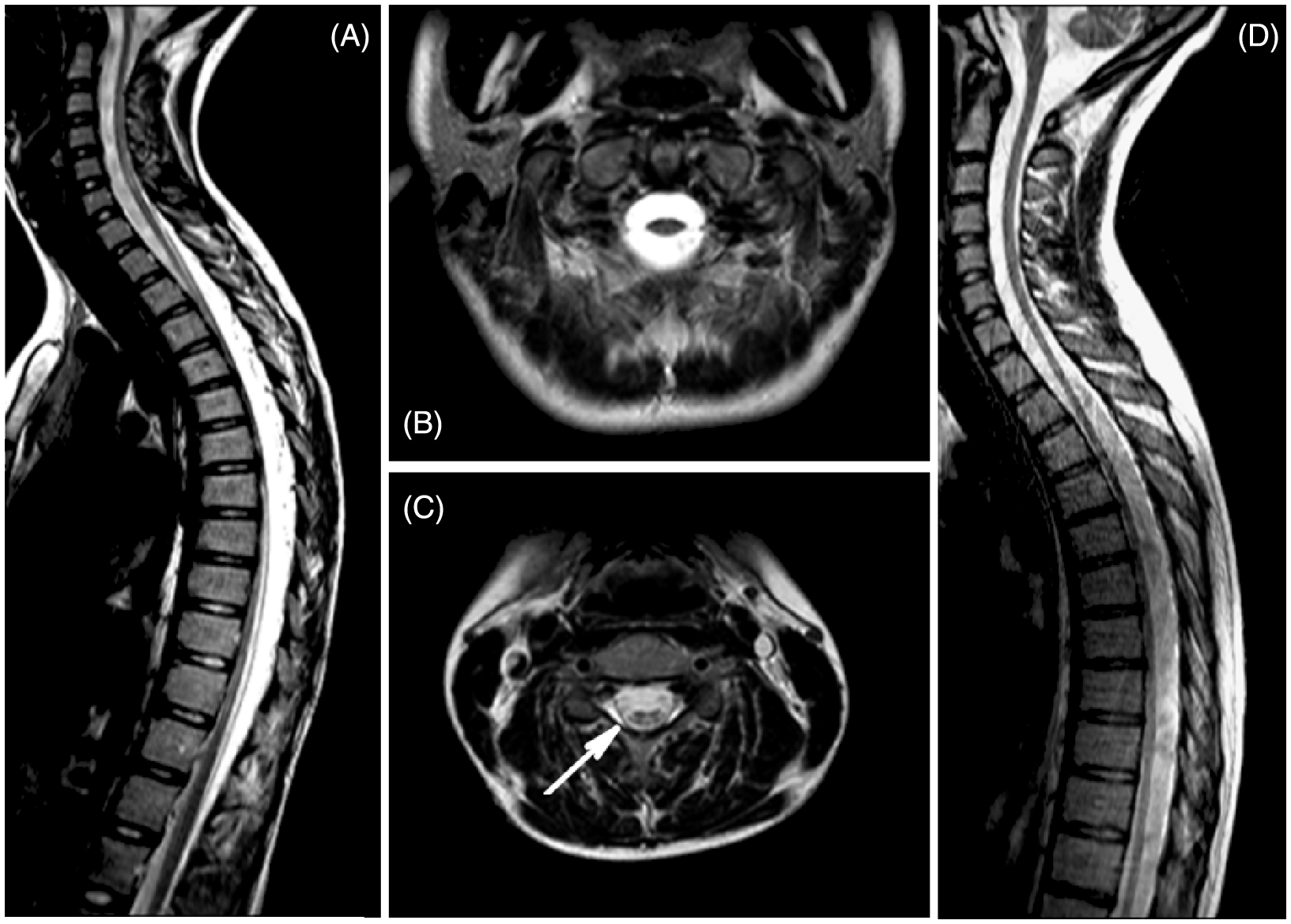
Cardiac and ophthalmology exams were normal. Tilt table testing was performed with a baseline heart rate (HR) of 60 bpm, and blood pressure (BP) of 105/62 mmHg. Head-up-tilting to 70° resulted in an increase in HR to 106 bpm and BP to 120/70 mmHg, and a syncopal episode at 7 min upright with dramatic drop of HR and BP. She recovered to baseline after 5 min supine. MR of her brain at age 14 showed T2 (T2weighted images on MRI) signal hyperintensity in the pons and external capsules (Figure 1A, B). On repeat imaging 2 years later the same observations were present, with additional evidence of volume loss in the pons. Spinal cord imaging also showed marked loss of volume throughout the entire spinal cord with evidence of hyperintense tracks (Figure 2A–C). MR spectroscopy with the first imaging revealed elevated choline and relatively low creatine within the abnormal pons. None of the imaging studies were performed during a migraine attack.
Abnormal increased T2 (T2 weighted images on MRI) signal in the pons and increased T2 signal intensity in the external capsules (A, B, case 1; C, D, case 2). Sagittal T2 images of the spinal cord showing diffuse cord atrophy (A, case 1; D, case 2). (B, C) Case 1 axial T2 images through the cervical spinal cord C1 level (B) and C4 level (C), showing marked atrophy. The spinal cord at C4 (C) shows regions of hyperintensity.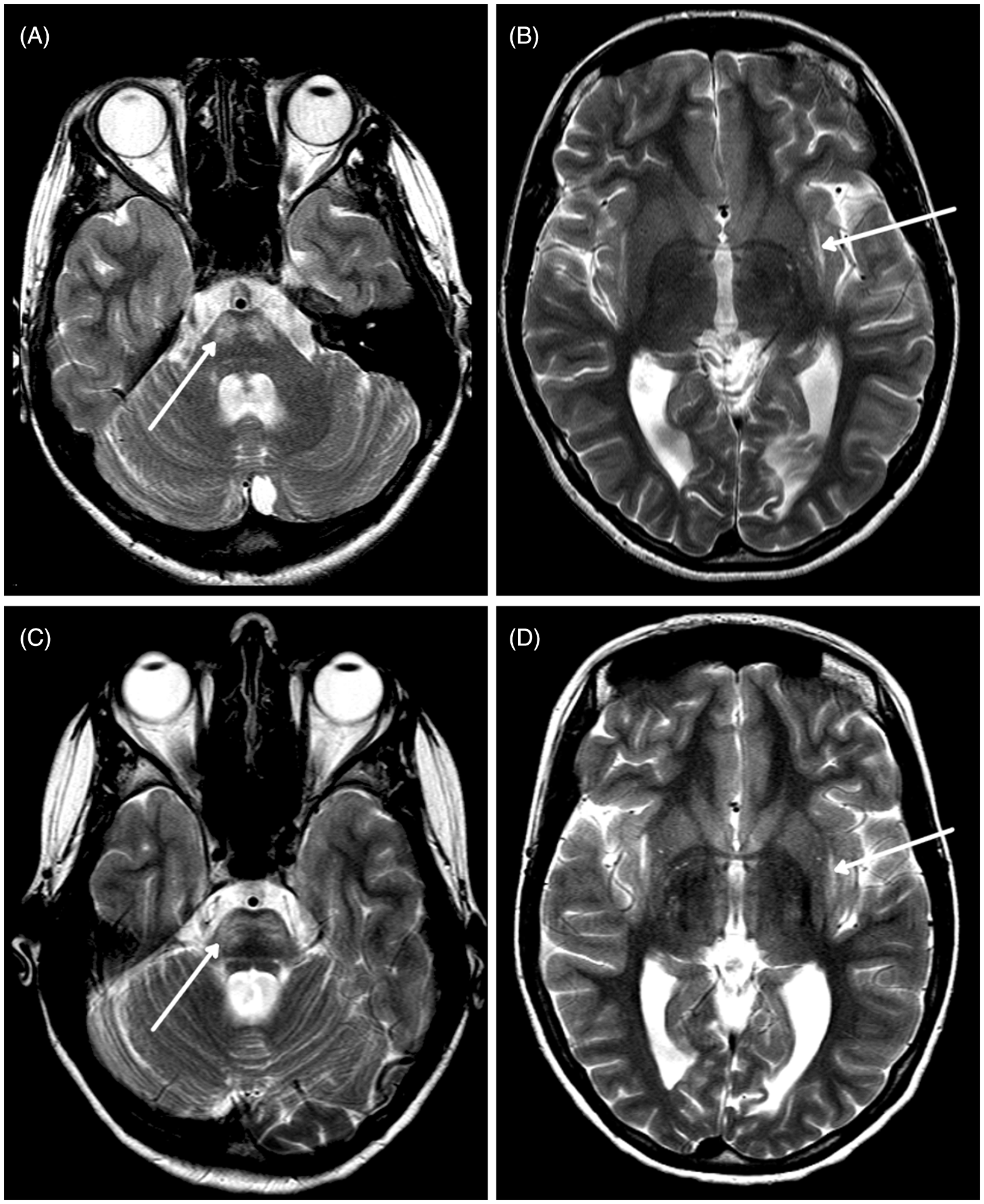
Analysis of CSF neurotransmitters and related metabolites was performed on two occasions, at ages 14 and 15. This analysis is included in our department in the evaluation protocol of most children with neurological presentations who undergo a lumbar puncture. 5-hydroxyindoleacetic acid (5HIAA, an end metabolite of serotonin) levels were very low on both occasions. Results for the other neurotransmitters, including dopamine and related metabolites, were unremarkable. CSF tryptophan levels were normal, which argues against activation of the kynurenine pathway with formation of the neurotoxin quinolinic acid (4).
We started treatment with 5-hydroxytryptophan (5HTP), the immediate precursor of serotonin, and carbidopa (to block the peripheral decarboxylase activity) at the age of 16.5 years (125 mg 5HTP and 25 mg carbidopa three times daily). Folinic acid (5 mg) and vitamin B6 (25 mg) daily were added. No adverse effects were observed with treatment. Additional medications included melatonin, gabapentin, oxybutynin, vitamin D and calcium.
Treatment resulted in clear improvement of ambulation and strength, as well as improvement in intensity and frequency of migraine, irritable bowel syndrome, speech, mood, sleep and limb weakness. Although before treatment she could not walk without assistance, after 8 months on therapy she could walk short distances without her crutches. After 3 years on treatment, she could walk without assistance short distances in the house, used her crutches at school, and used her wheelchair outside. She developed good upper body strength pushing the wheelchair on her own. Instead of biweekly episodes of migraine before treatment, she had about one episode every 2–3 months, lasting less than 24 hours. Involuntary withdrawal of 5HTP treatment for 3 days resulted in dramatic weakness and inability to hold a cup of tea, leading to severe burns of her thighs. The most recent prolonged episode of coma was the first episode of deterioration after a period of being stable.
Family history is positive for headaches in mother and maternal grandfather, and seizures in a paternal cousin. Her older sister (case 2) has similar but milder presentation. Another older sister and a younger brother are unaffected.
Case 2
The older sister originally presented at the age of 15 years with a history of hemiplegic migraine and seizures and myoclonic jerks. Her EEG showed generalized spike-and-wave activity, and polyspikes with photoconvulsive response, in keeping with juvenile myoclonic epilepsy. Her seizures were brief and infrequent and not associated with the migraine episodes. She did not have generalized tonic-clonic seizures. She subsequently developed progressive weakness, frequent falls, depression, and mild bladder instability. Her myoclonic jerks and seizures were well controlled with valproic acid. Imaging of the brain also showed band-like areas of increased T2 signal intensity in the pons as well as in the external capsules, as seen in case 1 (Figure 1C, D). There was also milder generalized atrophy of the spinal cord (Figure 2D).
CSF neurotransmitter analysis was performed at the age of 17 and revealed a low level of 5HIAA, homovanillic acid and normal tryptophan.
Treatment with 50 mg 5HTP and 12.5 mg carbidopa three times daily was started at the age of 17. She reported improvement in her ability to climb stairs and walk uphill after 6 months of treatment. At the age of 22 years she remains ambulatory. Her migraine has improved in frequency and she only has headaches without hemiplegia. Her depression and sleep have improved.
Methods
The lumbar punctures for CSF collection were performed in a fasted state, without sedation, none of them during a migraine attack, and all before the start of 5HTP replacement therapy. Fractions were collected in separate tubes according to protocol, immediately frozen, and analysed according to established methods at Medical Neurogenetics Laboratory in Atlanta, GA (5).
Platelet serotonin levels were measured in whole blood samples in a commercial laboratory.
Sequencing of the TPH2 and SLC6A4 genes performed in our research laboratory revealed no pathogenic mutations in coding exons or exon/intron boundaries. Both sisters were homozygous for the long 5HTTLRP allele in the SLC6A4 promoter region, and the 12 VNTR allele in intron 2, both variants being associated with higher expression of this gene. Ex vivo platelet serotonin uptake mediated by SERT was calculated using published methods (6). Platelet-rich plasma was prepared from fresh samples and diluted to a concentration of 2 × 105 cells per µl with autologous platelet-free plasma. Serial dilutions with 3H-labeled serotonin (specific activity 28.1 Ci/mmol) were made, between 10−8 M and 10−6 M and, after a 5 min incubation at 37°C, total and platelet3H-serotonin activity was measured. Separation of platelets was done with centrifugation.
The two migraine related genes, CACNA1A and ATP1A2, were analysed by full exon and intron/exon junction sequencing at the Medical Neurogenetics Laboratory in Atlanta, GA, USA. The CACNA1A sequencing included the newly identified exons 48a and 48b. The promoter region was assessed for the presence or absence of the 5-nucleotide deletion, the only reported pathogenic mutation in this region. MELAS molecular testing was performed in DNA extracted from muscle in the Clinical Biochemical Genetics Laboratory in our hospital.
Results
Neurotransmitters measured on two separate occasions in case 1 were: 5HIAA 33 nmol/l and 25 nmol/l respectively (reference 67–189), CSF tryptophan 2.69 µmol/l (reference: 1.26–3.8); case 2: 5HIAA 14 nmol/l (reference 67–189), HVA 105 nmol/l (reference 145–324), CSF tryptophan 2.66 µmol/l (reference 1.26–3.8).
Platelet serotonin levels were low in both patients: 18.3 and 27.1 ng per 1010 cells in case 1 and case 2, respectively (reference: 65–550 ng per 1010 cells), whereas values were within normal range in unaffected family members and control samples. Platelet serotonin levels increased to 39.8 and 48.6 ng per 1010 cells after 12 and 24 months of treatment in case 1 and to 37.9 ng per 1010 cells in case 2.
Platelet serotonin uptake was diminished (Figure 3). Vmax for case 1 was 30–50% of lowest control value, while Vmax for case 2 was at the low end of the control range. The Km for serotonin uptake was not affected.
Platelet serotonin uptake study results. Control data were generated from seven different controls, each assay performed in duplicate. The error bars denote the observed range of control values. Case 1 had uptake studies performed on two occasions (1a,1b), and case 2 had one assay performed. Platelets with added sertraline (a selective serotonin reuptake inhibitor) were used as a control demonstrating the specificity of the assay for serotonin transporter-mediated uptake of serotonin. S is serial concentrations between 10−8 and 10−6 and V is platelet serotonin uptake.
Discussion
Familial hemiplegic migraine (FHM) is a rare autosomal dominant subtype of migraine with aura in which attacks are associated with hemiparesis. FHM type 1 has been associated with mutations in the gene encoding the Cav2.1 α1 subunit of P/Q type Ca2+channels (CACNA1A) (7,8), and type 2 has been associated with mutations in the ATP1A2 gene, encoding a Na+/K+-ATPase subunit.
Delayed cerebral edema and coma has been reported after minor head trauma in FHM caused by the S218L mutation in the CACNA1A gene, and with the G301R mutation in the ATP1A2 gene (9–11). There are also reported cerebellar signs with these mutations, with ataxia, nystagmus, dysarthria, and cerebellar atrophy on imaging. Neither of our patients have cerebellar signs except the onset of dysarthria after the episode of coma in case 1. She did not have evidence of head trauma or any other obvious trigger before onset of her coma, and no pathogenic mutations were found with conventional sequencing in these migraine-related genes. In our patients the markedly low CSF 5HIAA and low platelet (plasma) serotonin levels are new findings, not previously described, and they indicate systemic serotonin deficiency, suggesting that some of the patients’ clinical manifestations might be associated with a defect in serotonin metabolism, clearance or release (12,13).
Serotonin has an important role in vascular resistance and blood pressure control. Its role in cardiovascular regulation includes two levels of control, one on a hormonal, vasoactive level, and one on a neuronal level, involving reflex reactions via sympathetic and parasympathetic pathways (14). This could explain the orthostatic intolerance found in case 1, with initially increasing heart rate and lower limb pooling followed by the orthostatic hypotension.
A functional serotonin transporter polymorphism has been associated with migraine (15,16). Since cortical spreading depression (CSD) was discovered by Leao in 1943, the relationship between CSD and migraine has been studied intensively. The effect of low serotonin on development of CSD was first reported in mouse studies in 2006 (17), when experimental serotonin depletion induced a characteristic repeated pattern of cortical depolarization. Using this model the hemiplegic migraine in our patients could be explained by the low systemic serotonin causing CSD.
Defects in the serotonin signaling contribute to the pathophysiology of irritable bowel syndrome (IBS) (18), and motor neurons in the Onuf nucleus have dense populations of serotonergic terminals facilitating sphincter contraction, possibly explaining the IBS and bladder instability in our patients. Serotonin is present in the anterior horn motor neurons and in the lateral part of the substantia gelatinosa, where it is associated with terminals of descending fibers (19), modulating segmental reflexes and nociception. The myelopathy and spinal cord atrophy identified in our patients still cannot be explained only by serotonin deficiency. Serotonin has also been associated with mood and sleep disorders, and also acts as a physiological regulator of lactation and involution and has a role in tumor-suppressing signaling in early stage breast cancers (20). Thus we suggest that the majority of the symptom complex identified in this family could be explained as a result of serotonin deficiency.
Possible underlying causes of the observed serotonin deficiency could include a defect in serotonin synthesis caused by TPH1 or TPH2 deficiency or decreased serotonin uptake caused by dysfunction of SERT. However, TPH1 is not expressed in brain, and we did not find a defect in TPH2 with sequencing. Adult SERT knockout mice have marked reduction (60–80%) in serotonin concentration in several brain regions, suggesting that the presence of a functional transporter is essential for brain serotonin homeostasis (21). Long-term SERT dysfunction causes serotonin depletion through negative feedback.
Although sequencing of SLC6A4 (encoding SERT) did not show pathogenic mutations, impaired SERT activity caused by disrupted posttranslational regulation of the transporter remains a possible mechanism for the patients’ hyposerotoninemia. The phenotype of SERT knockout mice (SERT−/−), including bladder dysfunction, disrupted somatosensory systems, sleep disorder, and gut hypermotility (IBS) (22), strikingly resembles the clinical syndrome observed in our patients. SERT knockout mice exhibit 50% reduction in serotonergic cell number and a fourfold decrease in firing rate in the dorsal Raphe nuclei. Biochemically the SERT knockout mice have markedly depleted serotonin tissue stores (23) and substantial depletion of neuronal serotonin and 5HIAA.
Some of the clinical symptoms, such as the strength, frequency and intensity of the migraine (except the recent episode of coma in case 1), and the mood and sleep improvement on serotonin replacement therapy, suggest that the systemic serotonin deficiency in these patients could be contributing to their neurodegenerative disorder. Platelet serotonin levels increased initially but did not reach the reference range, further supporting our hypothesis of SERT dysfunction as an important factor in the hyposerotoninemia of our patients.
Other cases with low CSF 5HIAA and positive response to treatment with 5HTP have been described (24,25). The underlying gene defect remained unidentified in these cases. None of the described patients had FHM. We hypothesize that the defect in SLC6 protein regulation observed in our patients may be secondary to an underlying cause that also affects other membrane proteins, such as the neuronal calcium channel or the Na+/K+ pump. Thus far we have not been able to identify the underlying genetic defect, but ongoing investigations including SERT expression studies and exome sequencing with bioinformatic approaches will yield more insights into the molecular cause and pathophysiology of this new disorder.
Footnotes
Funding
This work was supported by a Clinician Investigator Award from the University of British Columbia.