
Abstract
Background: Cluster headache (CH) is a rare cause of headache in children. Onset before 12 years of age is unusual, and long-term follow-up of pediatric cases has been not reported.
Objectives: To report three cases of CH with onset at childhood and at least ten years of follow-up.
Methods: Case report.
Results: The first case is that of a 12-year-old boy with episodic CH with unilateral pain and striking, bilateral autonomic manifestations, remitted for over eight years. The second case is unique in that it reports a case of chronic CH in a 13-year-old boy with Down syndrome. The third case is that of a 9-year-old girl with episodic CH with remissions of 2 and 5 years. All cases had prominent autonomic features. The frequency and duration of the attacks were similar to those that have been reported in adults. Good response to indomethacin was obtained in two cases, although tolerability issues occurred in one.
Conclusion: Sustained, long-term, medical and/or spontaneous remission occurs in CH of early onset. The phenotype and response to therapy in children, at least in these case examples, are similar to equivalent observations in adult patients with CH.
Keywords
Introduction
The Second Edition of the International Classification of Headache Disorders (ICHD-II) (1) describes 14 headache categories, sub-divided into a total of 196 possible headache diagnoses, 113 of which have been described in children. Although the diagnosis of the most common, primary headaches in children (e.g. migraine and tension-type headache) do not pose particular dilemmas to the clinician, several other headache forms are challenging to diagnose and treat. This complexity is aggravated by the fact that the phenotype of particular syndromes vary with age, and children often have verbal limitations to describe important clinical features required for diagnosis (2,3).
CH is rare in children, and the diagnosis of CH in children is often misleading because its incidence peaks between the ages of 20 and 40. Only about 5–10% of CH cases started at adolescence, and onset in childhood is very rare (4,5). In a multi-center pediatric study only two cases of CH were seen among 6629 children and adolescents attended in 27 Italian headache centers (6). Nonetheless, an autosomal dominant pattern is reported in about 5% of cases, and first degree relatives of CH probands have a 14-fold increased risk of being affected (7).
A recent systematic review found that only 80 cases of CH in children have been reported (6,8,9–13), and many of them did not meet the full ICHD-II diagnostic criteria for CH. In many instances, pain was described as bilateral, while others responded to propranolol or other migraine preventive medications, raising the question of whether CH has a different phenotype in children or whether there are diagnostic issues. Furthermore, of these cases, very few had onset in the first decade. Accordingly, the aim of this paper is to report three cases of CH in children followed for more than a decade, providing insights on the natural history and evolution of CH in children.
Case 1
This 12-year-old boy was first seen by one of the authors (MAA) in October 1999. Four months before consultation, the patient first experienced nocturnal attacks of the left side, severe periorbital pain accompanied by ipsilateral eyelid edema and conjunctival injection. The frequency gradually increased to 1–2 attacks per day, lasting between 60 and 90 min. Attacks recurred at similar times, and diurnal attacks were of lower intensity than nocturnal attacks. He was seen by an ophthalmologist, who recorded a normal intraocular pressure and diagnosed conjunctivitis. Despite topical antibiotic therapy the attacks continued with the same characteristics. After 3 months from onset, the patient was seen by a neurologist. Oxygen inhalation (7 l/min) was recommended, successfully aborting attacks within a few minutes. The patient's past medical history was unremarkable, and he had a positive family history for migraine (father, mother, and a cousin). Physical and neurological examinations were normal except for a slight reduction of the left side palpebral cleft. Magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) were normal. Verapamil (40 mg twice daily) and prednisolone (20 mg once daily) were started, and attacks progressively remitted, although verapamil had to be withdrawn after 5 days because of asthenia. Pain recurred at every attempt to taper down prednisolone. Methysergide (1 mg once daily), valproic acid (500 mg twice daily), and lithium (300 mg twice daily) were of limited efficacy. Meanwhile, the parents observed attacks with bilateral autonomic manifestations, one of them documented by MAA (Figure 1). In these ‘bilateral’ attacks, the pain was unilateral and still side-locked, but the eyelid edema and dilatation of peripheral conjunctival vessels were bilateral. After 5 months of taking prednisolone (20 mg once daily), the patient developed fever, and serological tests confirmed toxoplasmosis. Prednisolone was withdrawn, and indomethacin was introduced with total control of pain. After 1 month, indomethacin was discontinued, and attacks did not recur for 4 months. Indomethacin was re-introduced with total control of the attacks. Owing to dyspepsia, indomethacin was again discontinued. The pain attacks immediately recurred, and melatonin (3 mg twice daily) was introduced with total attack remission after 2 weeks of treatment; the drug was withdrawn after 1 month with success. After a remission period of 3 months, attacks recurred and were again controlled with melatonin. The patient had two other remissions lasting 6–19 months, and the last episode was controlled only with topiramate (100 mg twice daily). Topiramate was withdrawn after 4 months, and the patient has been CH free, without medication, for over 8 years.
Case 1. (A) Outside the cluster period; (B, C) during an attack, the bilateral eyelid edema (B), and bilateral dilatation of conjunctival vessels (C). Reproduced with permission.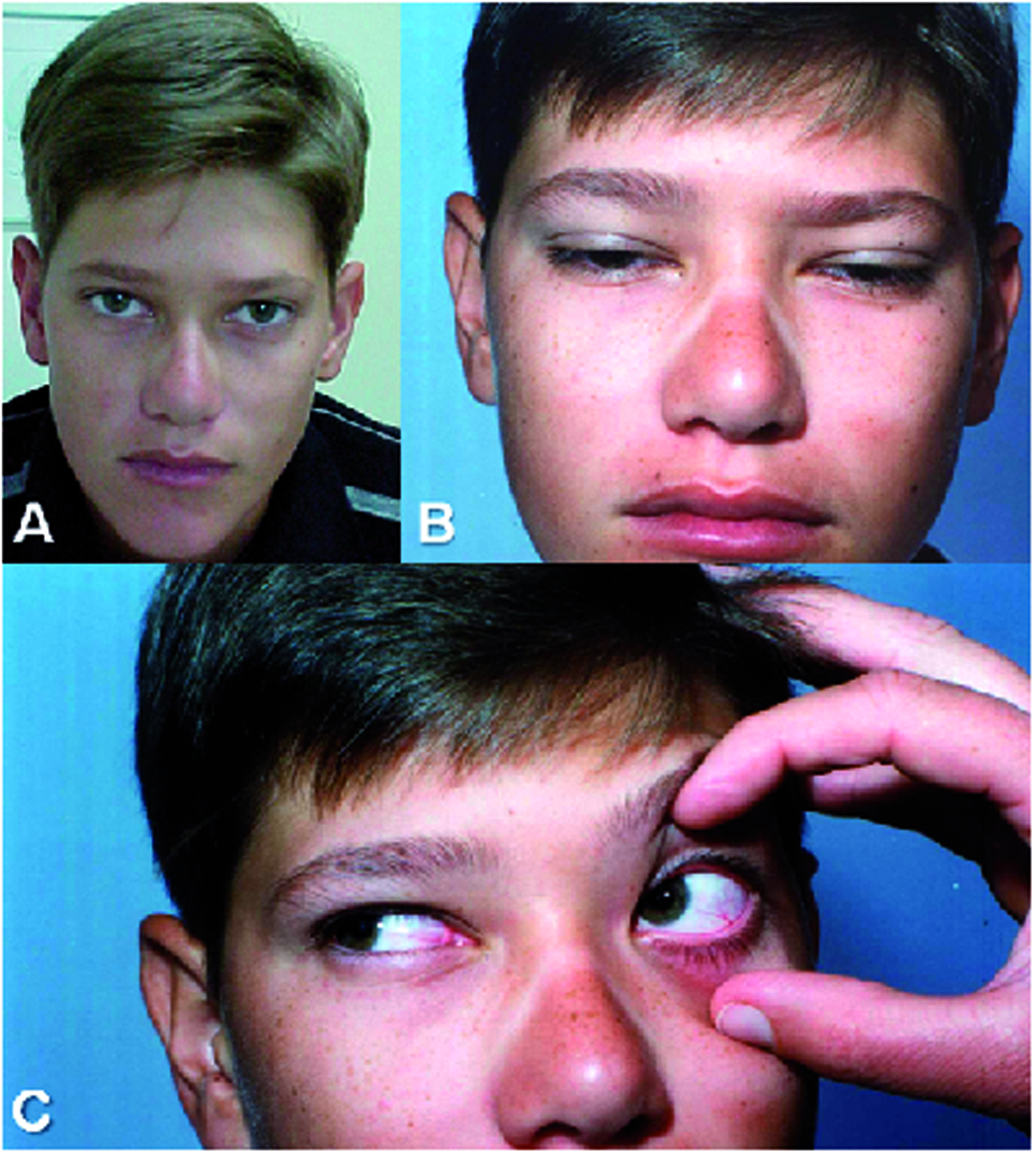
Case 2
In July of 2000, a 13-year-old boy with Down syndrome developed nocturnal episodes of excruciating headaches. His mother was sure that pain was unilateral, side-locked (left periorbital area), and accompanied by ipsilateral lacrimation and eyelid edema. The frequency gradually increased to 1–2 attacks per day, lasting 40–90 min. Attacks were nocturnal and only occasionally occurred in the early afternoon. He was referred to a neurologist, and a work-up included EEG, CT scan and MRI, all normal. A depot betamethasone injection (betamethasone dipropionate 5 mg and betamethasone disodium phosphate 2 mg) was made intramuscularly with total remission of the attacks for a period of 5 days. A subsequent attack was witnessed by a nurse who reported ipsilateral lacrimation, conjunctival injection and eyelid edema. When he was first seen by one of the authors (LB) in August 2000, a short course of prednisone was started but, after a few days, was interrupted because of intense myalgia. Attacks immediately recurred. The patient did not tolerate verapamil (asthenia). Lithium was started with total control of the attacks at 600 mg/day and blood level of 0.8 mEq/l. After 6 months, the patient was asymptomatic, and the lithium was tapered off with recurrence of attacks. He has been followed for nearly 11 years with total control of CH with lithium, and has suffered recurrence of attacks at every attempt of its discontinuation.
Case 3
A 9-year-old girl was first seen at the end of 2000, due to excruciating attacks of pain located in her right eye and periorbital area. Attacks lasted 40–60 min, and the frequency increased over 2 months from occurrences every other day to two attacks per day. She described the attacks as follows: ‘I can't remain seated. I want to tear my eye and hit my head on the wall.’ Her mother reported conjunctival injection, lacrimation, rhinorrhea and facial sweating at the right side during attacks. She was submitted to otorhinolaryngological and ophthalmological evaluations, and no abnormalities were found. Neuroimaging was normal. Dihydroergotamine was tried without success, and indomethacin was not tolerated. Three months after onset, methysergide was introduced with total control of the attacks. Methysergide was withdrawn after 9 months, and the pain attacks did not recur. After 1 year of remission, the attacks restarted with the same characteristics. Atthis time prednisolone and divalproex were started with total control of the attacks. Divalproex was discontinued after 3 months. After 5 years of remission, attacks recurred with the same characteristics, but no response with divalproex was obtained. Topiramate (50 mg twice daily) and prednisolone (40 mg once daily) were introduced with total control of the attacks after two weeks. Prednisolone was withdrawn after 2weeks, and topiramate after 1 month, with no recurrence of the attacks. The follow-up was discontinued 3years ago.
Discussion
This report discusses long-term series of CH in children and adolescents in three cases. According to the ICHD-II, two patients had episodic CH (Cases 1 and 3), and one child with Down syndrome had chronic, unremitting CH (Case 2). The longest period of remission was observed in Case 1 (the patient has been asymptomatic for the past 11 years).
Certain peculiarities of CH in children have been sparsely suggested in the literature and include: 1) A male/female proportion that seems to be lower than that commonly seen in adults (14); 2) shorter duration of cluster periods (14); 3) pain free intervals (of days) within the cluster period (14); 4) good response to indomethacin (15); and 5) less prominent autonomic manifestations (14). All cases had prominent autonomic features. Frequency of attacks was similar to what has been reported in adults. Autonomic manifestations (but not pain) were bilateral in one case. This pattern of unilateral pain and bilateral, autonomic symptoms has been rarely reported (16–18). Good response to indomethacin was obtained in two cases, although tolerability issues occurred in one.
Case 1 has interesting characteristics, and pharmacological control was difficult to obtain. Melatonin, a preventive medication of level C advice in the treatment of cluster headache (19), yielded total control. Once total control was obtained, the medication was discontinued, and CH has not recurred. Single episodes of CH are not common (5) and may be secondary to an identifiable insult.
Also noteworthy in case 1was the pattern of side-locked pain and bilateral autonomic symptoms. As established, autonomic signs in CH are usually reported as confined to the side of the pain; however, some studies have shown evidence of increases in lacrimation, nasal secretion, conjunctival injection, and ocular, sympathetic dysfunction occurring bilaterally (16,20,21). Barón et al. have documented bilateral, conjunctival injection during cluster headache attacks with unilateral pain in eight of nine consecutive adult patients (21). The conjunctival injection was predominantly on the side of the pain and involved the peripheral territory of the bulbar and tarsal conjunctiva but not the ciliary vessels. Such pattern of conjunctival involvement was observed in Case 1 (Figure 1) and may have clinical importance for differentiating CH attacks from pain that is secondary to glaucoma, uveitis, or keratitis, which are usually accompanied by pericorneal injection (21).
The second case is unique in that it reports a case of chronic CH in a child with Down syndrome. Down syndrome is sometimes associated with postural headaches due to intracranial hypotension, pseudotumor, or headache associated with sleep apnea (17, 18). To the best of our knowledge, CH has not been reported in association with Down syndrome. Because the use of steroids is problematic in children with this syndrome, treatment is not easy. Fortunately, we obtained total control with lithium.
Although the close temporal relationship between medication onset and CH remission suggest a causal relationship, it cannot be ruled out that the patients had spontaneous remissions. Nonetheless, since the aim of our report was to describe the long-term evolution of cluster headaches beginning at childhood, we think that the message is still valid. Medication-induced or spontaneous, prolonged remission was seen in all three cases.
Because CH is rare in childhood, identifying peculiarities is difficult. Although others have reported that CH attacks are of shorter duration and lower frequency in children than in adults, we have not seen evidence of this. It does seem that indomethacin is a good therapeutic option in children. Difficulty in gaining initial control does not seem to predict outcome.
Summary of the Cluster headache characteristics of the three reported cases

I = ineffective; NT = not tolerated; E = effective.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflicts of interest
MEB is a full-time employee of Merck Research Laboratories. He owns stocks and stock options of Merck. Merck did not sponsor this study. The other authors report no disclosures.