
Abstract
Background: This study evaluated the CGRP receptor antagonist MK-3207 for acute treatment of migraine.
Methods: Multicenter, double-blind, randomized, placebo-controlled, parallel-group, two-stage adaptive study with two interim efficacy analyses to facilitate optimal dose selection. Migraine patients were initially randomized to MK-3207 2.5, 5, 10, 20, 50 and 100 mg or placebo to treat a moderate/severe migraine. One or more doses were to be discontinued based on the first interim analysis and a lower or higher dose could be added based on the second interim analysis. The primary endpoint was two-hour pain freedom.
Results: A total of 547 patients took study medication. After the first interim analysis, the two lowest MK-3207 doses (2.5, 5 mg) were identified as showing insufficient efficacy. Per the pre-specified adaptive design decision rule, only the 2.5-mg group was discontinued and the five highest doses (5, 10, 20, 50, 100 mg) were continued into the second stage. After the second interim efficacy analysis, a 200 mg dose was added due to insufficient efficacy at the top three (20, 50, 100 mg) doses. A positive dose-response trend was demonstrated when data were combined across all MK-3207 doses for two-hour pain freedom (p < .001). The pairwise difference versus placebo for two-hour pain freedom was significant for 200 mg (p < .001) and nominally significant for 100 mg and 10 mg (p < .05). The incidence of adverse events appeared comparable between active treatment groups and placebo, and did not appear to increase with increasing dose.
Conclusions: MK-3207 was effective and generally well tolerated in the acute treatment of migraine.
Introduction
Calcitonin gene-related peptide (CGRP) is a neuropeptide that has a key role in the pathophysiology of migraine (1,2). The CGRP receptor antagonists olcegepant (given intravenously) and telcagepant (given orally) have been demonstrated to be effective and generally well tolerated in the acute treatment of migraine (3–7). The CGRP receptor antagonists evaluated to date appear to have overall efficacy generally similar to that of the triptans and may offer another treatment option for patients who are not responsive to or are suboptimally responsive to triptans, who have side effects with triptans or for whom triptans are contraindicated (8). CGRP receptor antagonists lack direct vasoconstrictor activity and may offer, in theory, advantages over triptans, where the potential for vasoconstriction is a perceived risk and has resulted in warnings and contraindications in the product labels regarding use in patients with cardiac disease (9).
MK-3207 is a new, highly selective CGRP receptor antagonist that is approximately 40- to 65-fold more potent than telcagepant for the human CGRP receptor in vitro, based on radioligand binding and cell-based functional assays (10,11). Pharmacokinetic results indicate MK-3207 is rapidly absorbed (median Tmax ∼1–2 hours). The concentration-time profiles exhibited bi-exponential decay post-Cmax, with an apparent half-life of about three to six hours for the α phase, and nine to 15 hours for the β phase. It was hypothesized that the higher potency of MK-3207 might lead to greater efficacy relative to telcagepant and existing oral triptan treatments. The objective of this dose-finding study was primarily to characterize the dose response of oral MK-3207 over a range of doses for the acute treatment of migraine and to identify effective doses for further study in pivotal efficacy trials. The study used an adaptive design to efficiently assess a broad range of doses.
Methods
Patients
Patients were eligible for the study if they were 18–65 years of age, in good physical health, with a greater than one year history of migraine with or without aura as defined by International Headache Society criteria (12) that typically lasted 4–72 hours if untreated, and had two to eight moderate or severe migraine attacks per month in the two months prior to the screening visit. Patients taking migraine prevention medication were allowed to enter the study provided that the prescribed daily dose had not changed during the three months prior to screening. Patients were excluded if they had difficulty distinguishing their migraine attacks from tension-type headaches, had a history of predominantly mild migraine attacks or migraines that usually resolved spontaneously in less than two hours, had more than 15 headache days per month, had taken medication for acute headache on more than 10 days per month in any of the three months prior to screening, had basilar-type or hemiplegic migraine headache, or were more than 50 years old at age of migraine onset. Due to potential pharmacokinetic interactions with MK-3207, patients taking potent CYP3A4 inhibitors, CYP3A4 inducers or drugs with narrow therapeutic margins with theoretical potential for drug interactions in the CYP2C family (e.g. warfarin) within one month of the screening visit were ineligible, and these medications were not permitted during the study. Patients with a history of cardiovascular disorder in the last six months, and women who were pregnant, breastfeeding or planning to become pregnant in the next six months were excluded.
Standard protocol approvals, registrations, and patient consents
The study was conducted in accordance with principles of Good Clinical Practice and was monitored by representatives of the study sponsor (Merck & Co., Inc.). The study was approved by the ethical review committee for each site and each patient provided written informed consent. The trial was registered at ClinicalTrials.gov (NCT00712725). A Scientific Advisory Committee comprised of non-Merck and Merck scientists contributed to the development of the protocol, statistical analysis plan, analysis and interpretation of the data, and authoring of the manuscript.
Study design
This multicenter, randomized, double-blind, placebo-controlled, parallel-group study (Merck protocol 005) was conducted at 47 sites in the United States (19 sites), Canada (9 sites) and Europe (19 sites) between July 2008 and January 2009. A two-stage adaptive design, with two interim efficacy analyses, was employed to facilitate optimal dose selection. The first interim efficacy analysis was performed in order to drop the less-effective dose levels from further randomization in Stage 2. A second interim efficacy analysis was performed to test for futility and potentially add a dose group at either the low or high end of the dose range. Patients randomized to the added group were distributed among the existing sites. Any modifications made as a result of the interim analyses were performed in a blinded fashion and were not apparent to patients or investigators.
Patients were allocated using a computer-generated randomized allocation schedule prepared by a blinded statistician at Merck. Numbered containers were used to implement allocation. Personnel at each study site used a central interactive voice response system (IVRS) to determine which container should be given to which patient. Other than as noted below, study personnel, including investigators, study site personnel, patients and Merck staff remained blinded to treatment allocation throughout the study.
Stage 1
In Stage 1, patients were screened and randomized to MK-3207 or placebo. Approximately 312 patients were to be randomized to receive a single dose of MK-3207 2.5, 5, 10, 20, 50, or 100 mg (N = 39 for each dose), or matching placebo for each dose (total N = 78) in a double-dummy design. All patients took two tablets provided in separate bottles—depending on the dose strength, this could be two active tablets, two placebo tablets or one placebo and one active tablet; all patients took the same number of tablets of each size. Approximately 25% of all patients were to be randomized to placebo, and this ratio was to be maintained throughout the study. Patients were stratified according to their self-reported retrospective usual migraine response to a triptan. The two strata were: (1) triptan response ≥75% of the time; (2) triptan response < 75% of the time or triptan naive.
Patients were provided with a migraine diary to record their migraine experience before, during and following treatment for a single acute moderate-to-severe qualifying migraine attack. Patients were also instructed to call into an IVRS within 24 hours to record their rating of migraine intensity two hours after dosing with study medication.
The first interim analysis occurred after 312 patients had been randomized and a minimum of 20 patients had been treated in each of the three-lowest MK-3207 dose groups (the criteria for triggering this interim analysis were evaluated in an automated fashion within the IVRS). At the first interim efficacy analysis, the observed proportion of patients who obtained pain freedom at two hours for each of the six MK-3207 treatment groups was calculated from the patient-reported data within the IVRS by an unblinded statistician and compared to that of the combined placebo group.
The lowest MK-3207 group (“dose S”), up to 20 mg, with a two-hour pain free response that was at least 15 percentage points greater than that of the combined placebo group was identified. In order to increase the likelihood of observing a dose-response relationship from the study, a test of “poor dose response” was performed. If the two-hour pain-free response rate at any of the doses below dose S was at least 10 percentage points below the next-lower dose (i.e. poor dose response), all doses were to be continued into Stage 2. If a poor dose response was not identified (based on the above criteria), the MK-3207 dose below the lowest effective dose (“dose S-1”) and higher doses were to be continued into Stage 2. By the definition, a minimum of the four highest MK-3207 dose levels were to be continued into Stage 2, regardless of the outcome of the interim efficacy analysis.
Stage 2
In Stage 2, approximately 240 patients were to be randomly allocated to the MK-3207 dose levels remaining from Stage 1 (MK-3207 or placebo). For any continuing MK-3207 dose below dose S (the lowest identified promising dose), 18 patients were enrolled as well as six patients to each of the matching placebo groups. The remaining MK-3207 patients were equally allocated to the remaining higher doses. The randomization ratio of active MK-3207 to placebo remained at 3 : 1. This adaptive strategy had the potential to result in unequal numbers of patients in some of the lower MK-3207 dose groups, due either to discontinuation of one or more doses after the first interim efficacy analysis or the continuation of one or more doses below dose S into Stage 2, but with a reduced number of patients compared to the higher-dose groups.
The second interim analysis was triggered when a minimum of 40 patients were treated in each of the three highest MK-3207 dose groups, or a total of 460 patients had been randomized in the trial, whichever occurred first. The purpose of the second interim analysis was to determine whether a higher (200 mg) or lower (1 mg) dose of MK-3207 should be added. At this interim analysis, two separate evaluations were performed. The first was an assessment of futility, which was to determine whether the three top doses (20, 50, 100 mg) had suboptimal efficacy (thereby indicating a need to add the 200-mg arm to attempt to identify an effective dose). The second evaluation was an assessment of dose response and evaluation of a lack of sufficient separation of the lowest and highest doses on efficacy (thereby indicating a need to add the 1-mg arm to attempt to identify a minimally effective dose). Per the study design, either the 1-mg dose or the 200-mg dose could be added, not both.
The MK-3207 200-mg dose was to be added if the treatment gain over placebo in two-hour pain freedom in the three highest MK-3207 doses were all ≤15%. The MK-3207 1-mg dose was to be added if the treatment gain of MK-3207 2.5 mg over placebo in two-hour pain freedom was ≥20% but the difference between response rates for the MK-3207 100-mg and 2.5-mg arms was ≤5%. The added treatment group and matching placebo group were to enroll approximately 100 additional patients in a 3 : 1 ratio of active to matching placebo.
Procedure
Patients who met all the study entry criteria were enrolled and provided with study drug (MK-3207 or placebo) to be taken on an outpatient basis as soon as they experienced a moderate or severe migraine headache. Patients were instructed not to treat with rescue medication until after their two-hour pain assessment. If they were still experiencing a moderate or severe headache at two hours following initial treatment, they could take their own rescue medication.
During the 48 hours following the initial dose of study medication, patients recorded subjective assessments of pain severity, associated migraine symptoms and other measures at specified time intervals in a migraine diary. Patients also recorded information about any adverse events. Patients returned to the study site within approximately five to seven days after treatment to allow review of the diary, assessment of medication compliance, and tolerability and safety monitoring.
Assessments
Headache severity was assessed using a four-grade scale (no pain, mild pain, moderate pain, severe pain) at baseline (0 hour—time of taking study medication) and at 0.5, 1, 1.5, 2, 2.5, 24 and 48 hours post-dose. The presence or absence of phonophobia, photophobia, nausea, vomiting and ratings of functional disability (four-grade scale—normal, mildly impaired, severely impaired, requires bed rest) were recorded at the same time points as the headache severity ratings. For those patients who reported pain relief (reduction of pain to mild or none) or pain freedom (no pain) at two hours post-dose, the presence or absence of headache worsening (recurrence) within two to 24 hours and 24 to 48 hours was recorded. The use of rescue medication within 48 hours after dosing was also recorded. Patients also completed the Migraine Quality-of-Life Questionnaire (13) 24 and 48 hours after dosing and a Treatment Satisfaction Questionnaire for Medication assessment (14).
Tolerability and safety were assessed via spontaneous adverse event reports and routine pre- and post-study physical and laboratory examinations, electrocardiograms (ECG), and vital signs. Unblinded results from interim analyses were reviewed during the study by a data monitoring committee, comprised of individuals from Merck who were not directly involved with the study or the clinical development of MK-3207. The committee could make recommendations to terminate randomization to specific dose levels for safety reasons.
Statistical analysis
The primary efficacy variable was the proportion of patients reporting pain freedom at two hours post-dose. There were five secondary efficacy endpoints: (i) pain relief (reduction of moderate to severe migraine headache to mild or none at two hours; (ii) absence of photophobia at two hours; (iii) absence of phonophobia at two hours; (iv) absence of nausea at two hours; and (v) 2–24 hour sustained pain freedom (pain free from 2–24 hours without the use of rescue medication). The exploratory endpoint of 2–48 hour sustained pain freedom was also evaluated, as there has been a suggestion in previous studies that CGRP receptor antagonists might result in improved sustained response. Other exploratory efficacy endpoints were prespecified for analysis (e.g. functional disability, quality of life, satisfaction with medication) but are not included in the present report, which focuses on the primary and secondary endpoints.
The primary hypothesis was that a positive response trend exists across MK-3207 doses, as measured by the percentage of patients who achieve two-hour pain freedom. The analyses of the dose-response trend were performed using a generalized linear (regression) model with categorical terms for baseline headache severity, stratum (usual triptan response) and geographic region, with treatment group and age also included as continuous covariates. An identity link was used in the model in order to estimate treatment effects in term of the proportion difference, which is traditionally used in the migraine area. Covariates were selected based on previous data from the telcagepant development program suggesting an influence on response (severity, age, usual triptan response), or standard convention (geographic region as a surrogate for study site). A linear contrast comparing the response percentages across all treatment groups was used to test the primary efficacy hypothesis and similar hypotheses on the secondary endpoints.
In addition, it was hypothesized that at least one MK-3207 dose is superior to placebo in the treatment of acute migraine, as measured by the percentage of patients who achieve two-hour pain freedom. The pairwise comparisons of response percentages were performed using the same generalized linear regression model. Comparisons between treatment groups were made by the appropriate contrasts from the model. In addition, the model was also used to estimate the treatment responses for individual treatment groups. In the analysis of response percentages, the dose-response trend test was the primary test and served as the gatekeeper for the subsequent pairwise tests between MK-3207 and placebo doses. Hochberg's step-up method was used to adjust nominal p values from the pairwise comparisons at doses S, previously defined as the lowest effective dose identified in Stage 1 and above. Only nominal p values were provided for descriptive purposes for pairwise comparisons below dose S.
The efficacy analysis was based on the Full Analysis Set (FAS) population, in which patients were counted according to the treatment group to which they were randomized. For each two-hour endpoint, the minimum requirement for inclusion in the FAS population was that patients had taken treatment and had both a baseline headache severity measurement and at least one post-dose efficacy measurement prior to, or including, the two-hour time point. Missing data for headache severity and associated symptoms in the FAS analysis were imputed by applying the “last observation carried forward” method. Baseline values were not “carried forward” to impute missing post-treatment data.
All patients who were randomized and treated with the study drug were included in the safety assessment. The analysis was based on the “all patients as treated” population, in which patients were counted in the treatment group for the drug they actually received rather than in the treatment group to which they were randomized. Safety and tolerability were assessed by review of all safety parameters, including adverse events, laboratory values, ECG and vital signs. All adverse events up to 14 days following the treatment were included in the safety analysis. Safety endpoints included the proportions of patients with the following adverse events: (i) at least one; (ii) drug-related; (iii) serious; (iv) triptan-related (chest pain, chest tightness, throat tightness, asthenia, paresthesia, dysesthesia, hyperesthesia). The following changes in laboratory test values during the study were pre-specified as events of clinical interest: alanine or aspartate aminotransferase elevations more than three times the upper limit of normal, and absolute lymphocyte count of <0.85 thou/MCL.
Power
An extensive computer simulation study was performed to determine the sample size and power of the study. The placebo response rate was assumed to be 10% and the highest dose-response rate was assumed to be 30% in the simulations. The null hypothesis (all dose responses are the same as placebo with a response rate of 10%) and alternative hypothesis scenarios in which different trends of increasing responses between 10% and 30% were studied. Based on the primary trend test statistic with a total randomized sample size of 552 in the MK-3207 active and placebo arms (excluding the potential additional 1-mg or 200-mg MK-3207 arm), and assuming the non-evaluable rate was no more than 25%, the resulting study power was at least 90%.
Results
Patient accounting and demographics
CONSORT study flow chart: values are number of patients

CONSORT = Consolidated Standards of Reporting Trials. Placebo is the pooled arm of all matching placebo doses.
Patients may have been excluded for more than one reason.
Includes two patients from the active treatment groups who actually took placebo, due to taking treatment from only one of the two study medication bottles that they were provided with (one contained active treatment and one contained placebo).
One patient actually took placebo, due to taking treatment from only one of the two study medication bottles that they were provided with (one contained active treatment and one contained placebo), so their data is counted in the placebo group for the safety analysis.
Patient demographics and characteristics of treated migraine attacks at baseline: values are number (%) of patients except for age, where the mean (SD) is given

SD = standard deviation.
Baseline = 0 hours, immediately before patient took study drug.
Efficacy
Summary of efficacy of treatment on primary and secondary endpoints

N = number of patients in endpoint-specific full analysis set (FAS) population for primary endpoint; numbers were similar for other endpoints. n = number of treatment responders for the specific endpoint.
CI = confidence interval. Placebo is the pooled arm of all matching placebo doses. *p ≤ 0.050, **p ≤ 0.010, ***p ≤ 0.001 for MK-3207 vs. placebo pairwise comparison based on the model adjusting for baseline headache severity, geographic region, age and usual triptan response.
Trend test is based on generalized linear regression model adjusting for geographic region, baseline severity, usual triptan response and age with continuous covariate for treatment.
The test of the primary hypothesis, that there is a positive response trend across MK-3207 doses as measured by the proportion of patients who achieved two-hour pain freedom, was statistically significant (p < .001) in the context of the generalized linear regression model, in which treatment group was included as a continuous covariate. The pairwise comparison of each of the active groups to the placebo group with respect to two-hour pain freedom was statistically significant for the 200-mg group (p < .001) after controlling for multiplicity across all Stage 2 doses, and was nominally significant for the 10-mg and 100-mg (p < .05) groups. A graphical representation of the results for the primary endpoint is provided in Figure 1. There was no effect of the stratification factor of usual migraine response to a triptan on the two-hour pain freedom response.
Observed percentage of migraineurs with pain freedom at two hours and associated 95% confidence intervals. p values shown are for pairwise comparisons with the placebo group based on the model adjusting for baseline headache severity, geographic region, age and usual triptan response.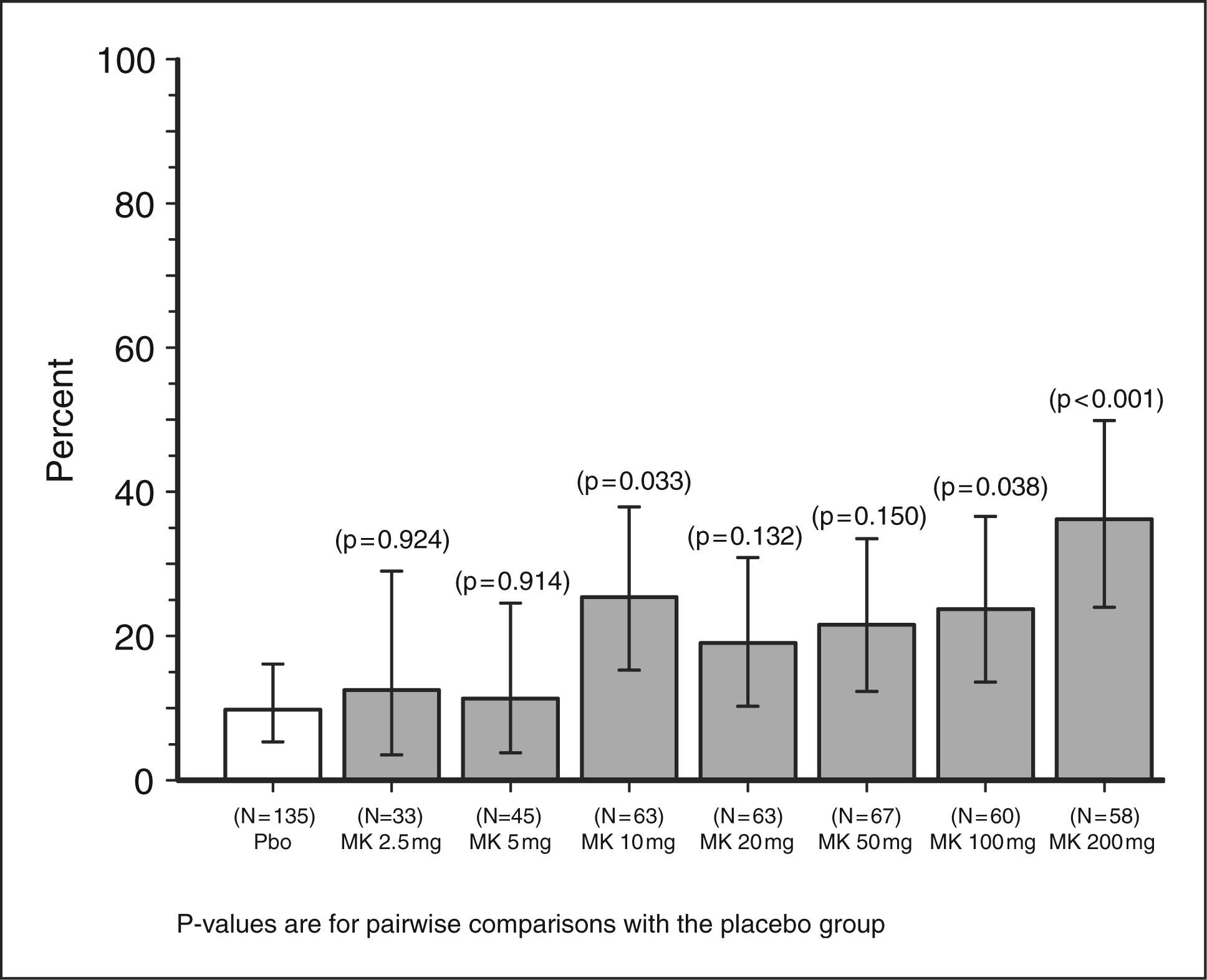
A statistically significant positive trend across MK-3207 doses was also seen for all secondary endpoints and the 2–48-hour sustained pain freedom exploratory endpoint (Table 3). For two-hour pain relief, the pairwise comparisons versus placebo were significant for all doses above 10 mg. For the remaining secondary endpoints of two-hour freedom from photophobia, phonophobia and nausea and 2–24-hour sustained pain freedom, as well as the 2–48-hour sustained pain freedom exploratory endpoint, only the 200-mg dose was consistently significantly more effective than placebo.
Tolerability and safety
Number (%) of patients reporting clinical adverse events within 14 days post-dose

N = patients who took at least one tablet of the study medication; placebo is the pooled arm of all matching placebo doses.
As determined by the investigator.
Triptan-related adverse events include chest pain, chest tightness, throat tightness, asthenia, paresthesia, dysesthesia or hyperesthesia
Incidence ≥5% in one or more treatment groups.
There were no deaths. There were three serious adverse events in two patients, which the investigator considered not related to study medication. One patient in the placebo treatment group experienced serious adverse events of congestive heart failure and hypersensitivity following a car accident. Another patient in the MK-3207 10-mg group was diagnosed with an ovarian cyst. All serious adverse events resolved.
Four events of clinical interest were reported. One patient assigned to the MK-3207 20-mg group experienced an event of increased aspartate aminotransferase to more than three times the upper limit of normal at the post-treatment visit (seven days after taking study treatment) and this was considered to be related to study medication by the investigator. Three patients had an event of decreased lymphocyte count of <0.85 thou/MCL. The patients were assigned to placebo, MK-3207 10-mg and the MK-3207 200-mg groups, respectively. Only the event in the placebo group was considered to be related to study medication by the investigator. The patient randomized to MK-3207 200-mg had a lymphocyte count <0.85 thou/MCL at baseline. In all cases, levels returned to normal at follow-up visits.
Laboratory abnormalities during the study were uncommon and no clinically relevant differences were seen between treatment and placebo groups. Other assessments including the percentage of patients who exceeded pre-defined levels of change on laboratory parameters, vital sign measurements, ECG measurements and physical examinations indicated no clinically meaningful differences between treatment groups.
Composite efficacy plus tolerability
An exploratory assessment was performed using a relatively new composite measure intended to summarize the overall clinical profile (efficacy plus tolerability) of treatment over the initial 24-hour period after dosing. The measure was the percentage of patients with 2–24-hour sustained pain freedom who also reported no adverse events from 0–24 hours (15). The results from this analysis suggested that the 200-mg dose appeared to have the most favorable clinical profile (Figure 2).
Observed percentage of migraineurs with 2–24 hour sustained pain freedom plus no adverse events and associated 95% confidence intervals. p values shown are for pairwise comparisons with the placebo group based on the model adjusting for baseline headache severity, geographic region, age and usual triptan response.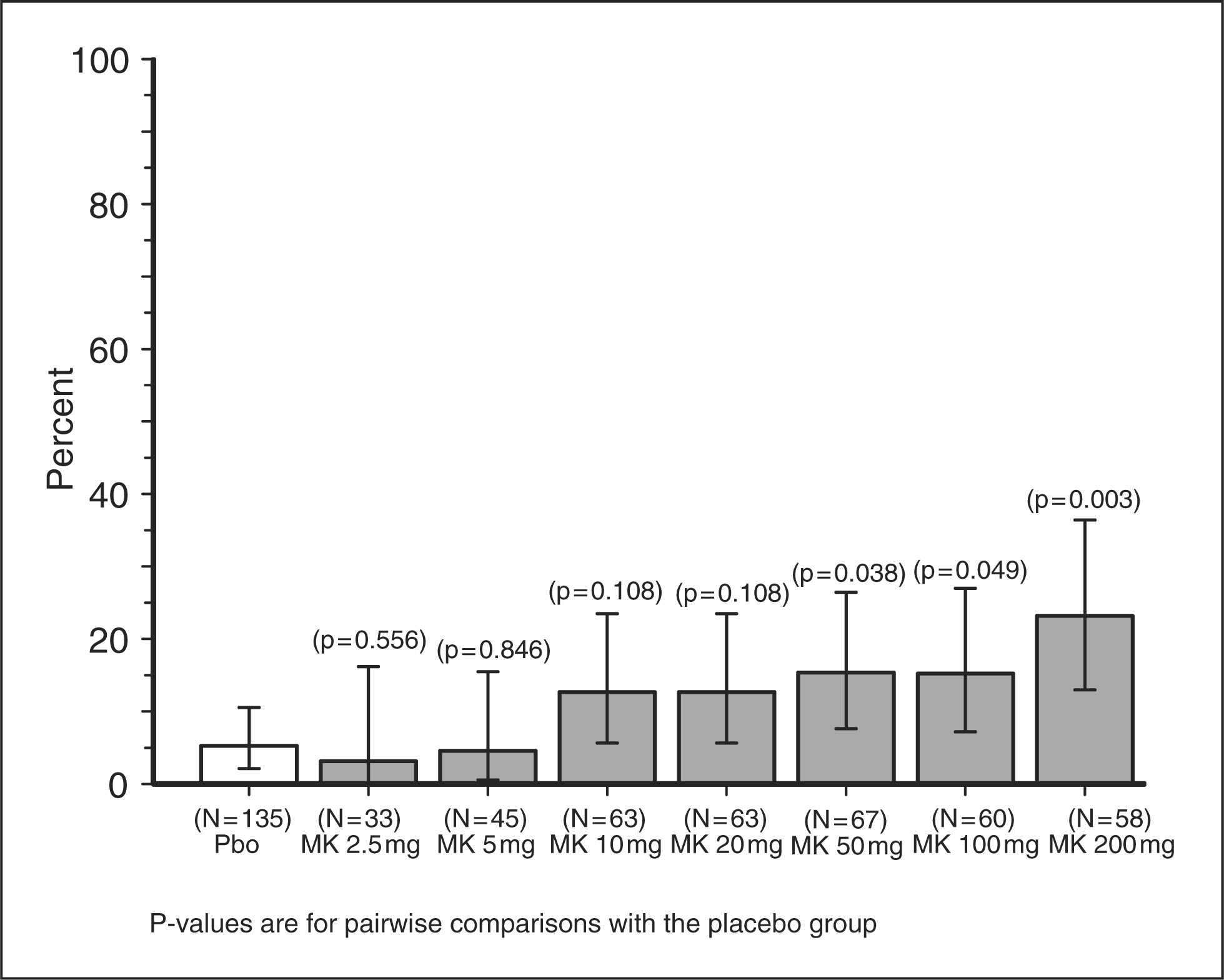
Discussion
Our findings support previous data indicating that CGRP receptor antagonists are effective in the acute treatment of migraine (3–7). A dose-response trend was observed for all primary and secondary efficacy endpoints, and the 200-mg dose appeared to be the most effective. There was no active comparator in our study, but based on historical comparison, the efficacy of the 200-mg dose appeared greater than that of triptans and other CGRP receptor antagonists in previous studies. For example, the placebo-subtracted two-hour pain freedom response rate was 26% for MK-3207 200 mg in the present study versus 20% observed for sumatriptan in a meta-analysis (16) and 17% seen for telcagepant 300 mg in a previous Phase 3 trial (5). However, an important limitation of the adaptive study design is that the relatively small sample sizes per dose imposes limits on the conclusions that can be drawn regarding the relative efficacy of individual doses. The confidence intervals for efficacy measures for each dose were overlapping, and it is not possible to definitively conclude that the 200-mg dose was more effective than other doses from 10 mg and up. It is possible that the apparent greater efficacy of the 200-mg dose was a chance finding, and that 10 mg represented the plateau of efficacy.
If, on the other hand, the apparent dose-response trend represents a true finding, then it is possible that doses higher than 200 mg might be more effective. The range of doses initially selected (2.5 mg, 5 mg, 10 mg, 20 mg, 50 mg and 100 mg) was based on the following rationale. Target engagement and identification of a potential dose range for CGRP receptor antagonism was previously demonstrated by using the capsaicin-induced dermal vasodilatation model in humans (17,18). Data from this model, which assesses inhibition of peripheral CGRP receptors, suggested an EC90 value of approximately 14 nM for inhibition of capsaicin-induced dermal vasodilatation. Based on this data and prior experience with the CGRP receptor antagonist telcagepant, it was anticipated that the clinically efficacious dose of MK-3207 would be between 2.5 mg and 100 mg; this dose range is associated with four-hour plasma concentrations of approximately 0.4 to 200 nM in healthy humans. One possible explanation for the suggestion in our study that the efficacy of MK-3207 appeared to be increased at a dose greater than that necessary to produce maximal inhibition of peripheral CGRP receptors in the capsaicin model is that additional central blockade of CGRP receptors may be important, and that high plasma levels are necessary to achieve central penetration. However, this is speculative at present.
In agreement with the previous findings using CGRP receptor antagonists, we found that MK-3207 was generally well tolerated for the acute treatment of migraine. The incidence of patients reporting adverse events within 14 days appeared comparable between active treatment groups and placebo, and there did not appear to be evidence of an increase in adverse events with increasing dose. Thus, on a composite measure of efficacy and tolerability over 24 hours (sustained pain freedom plus no adverse events) the differentiation between the 200-mg dose and the other doses was more apparent than when looking at efficacy measures alone. As was observed for telcagepant, the most common adverse events for MK-3207 were nausea, dizziness, fatigue, dry mouth and sleepiness. One patient assigned to the MK-3207 20-mg group experienced an adverse event of increased aspartate aminotransferase more than three times the upper limit of normal. This level returned to normal at the follow-up visit. In extended Phase 1 clinical pharmacology studies of MK-3207, some subjects experienced delayed liver-test abnormalities, generally following discontinuation of drug administration. Based on the Phase 1 information, it was decided to discontinue development of MK-3207 (19).
Study investigators
Belgium: Luc Herroelen, Nina De Klippel, Jean Schoenen, Michel Van Zandijcke. Canada: Werner John Becker, Guy Boudreau, Suzanne Christie, Marek Gawel, Rosella Giammarco, Jonathan Gladstone, Allan Purdy, Gary Shapero, Lucian Sitwell. Germany: Veit Becker, Hartmut Goebel, Arne May, Volker Pfaffenrath, Gunther Schumann. Italy: Anna Ambrosini, Piero Barbanti, Maria Adele Giamberardino, Paolo Martelletti, Luigi Alberto Pini. Sweden: Carl Dahlof, Elisabet Waldenlind, Lars Edvinsson, Boris Klanger, Mattias Linde. United States: Sheena K. Aurora, Gary Berman, Elizabeth M. Bretton, Bruce C. Gebhardt, Larry I. Gilderman, Daniel B. Groblewski, Daniel E. Grosz, Steven G. Hull, Stephen W. Kayota, David Ellison, Paul J. Markovitz, David S. Miller, Lawrence Robbins, Larry S. Seidman, Stuart Robert Stark, Michael M. Tuchman, Merle Lea Diamond, Mark Yen, Burton Ginsberg.
Competing interests
This study was funded by Merck Research Laboratories. D.J.H., Y.G., R.B., D.T., X.F., C.A., C.L., and T.W.H. are employees of Merck & Co., and own stock or stock options in Merck.
S.K.A. has received grant and research support from Advanced Bionics, Alexza, Allergan, GlaxoSmithKline, MAP Pharmaceuticals, Merck, Ortho-McNeil, Neuralieve and Takeda; has served as a consultant for Ortho-McNeil Pharmaceutical, Merck, GlaxoSmithKline, Allergan and Neuralieve; and has received honoraria from Merck, GlaxoSmithKline, NuPathe and Ortho-McNeil Pharmaceutical.
D.W.D. has received honoraria from and has had consulting agreements with Allergan, Pfizer, Merck, Endo, OrthoMcNeil, Coherex, MAP, Neuralieve, Addex, Solvay, Eli Lilly, Neuraxon, Minster, HS Lundbeck and Kowa, has research grants from AstraZeneca, Medtronic, St Jude and Advanced Neurostimulation Systems, and has independent support from the National Institute of Neurological Diseases and Stroke (NINDS) and the Mayo Clinic.
P.J.G. has consulted over the last 12 months for, advised or collaborated with Allergan, Almirall, Amgen, ATI, AstraZeneca, Belgian Research Council, Boehringer-Ingelheim, BMS, Boston Scientific, Colucid, Eli Lilly, Fidelity Foundation, GlaxoSmithKline, Johnson & Johnson, Kalypsys, Medtronic, MAP, Migraine Research Foundation, Migraine Trust, Minster, Medical Research Council-UK, MSD Pharma, NINDS, Netherlands Research Council, Neuralieve, Neuraxon, NTP, Organisation for Understanding Cluster Headache-UK and US, and Pfizer.
Footnotes
Acknowledgements
This study was funded by Merck & Co., Inc. The authors would like to thank Sheila Erespe from Merck for assistance in formatting the manuscript and Scott Grossman for helpful comments on a draft version.