
Abstract
Glutamatergic hyperactivity is implicated migraine pathogenesis. Also, LY293558, an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate (KA) receptor antagonist, is effective in preclinical models of migraine. We therefore tested LY293558 in acute migraine. We conducted a randomized, triple-blind, parallel-group, double-dummy, multicentre trial of 1.2 mg/kg intravenous (IV) LY293558, 6 mg subcutaneous (SC) sumatriptan, or placebo in the treatment of acute migraine. The primary efficacy variable was the headache response rate, i.e. headache score improvement from moderate/severe at baseline to mild/none at 2 h. Of 45 enrolled patients, 44 patients (20M:24F; mean age ± SD = 40 ± 9 years) completed the study. Response rates were 69% for LY293558 (P = 0.017 vs. placebo), 86% for sumatriptan (P < 0.01 vs. placebo) and 25% for placebo. LY293558 and sumatriptan were superior to placebo (P < 0.01 for all comparisons) on all other measures of improvement in pain and migraine associated symptoms. Fifteen percent of patients who took LY293558 reported adverse events (AEs) (n = 2; one mild, one severe). Fifty-three percent of patients who took sumatriptan (n = 8; seven mild, one moderate) and 31% of those who received placebo reported AEs (n = 5; four mild, one severe). The efficacy and safety results of LY293558 in this small migraine proof of concept trial, together with supportive preclinical data, provide evidence for a potential role of nonvasoactive AMPA/KA antagonists in treating migraine. Larger trials are needed to further test the hypothesis.
Introduction
There is a body of literature that supports a role of glutamate in the pathogenesis of acute migraine. Glutamate receptors mediate neuronal impulse trafficking in the trigeminal nucleus caudalis (TNC) (1), the primary relay station of pain processing from the head and face. Iontophoretic application of
The role of glutamate in migraine pathogenesis is further supported by studies that show elevated plasma levels of glutamic acid, glutamine, glycine, cysteic acid and homocysteic acid interictally and more so during an attack (9, 10). Furthermore, migraineurs exhibit signs that correspond to electrophysiological observations in animals consistent with central sensitization (11), a process of excessive activation of neurons in the dorsal root ganglia or the TNC following peripheral sensory stimulation (12), and which is mediated by glutamate receptor activation.
The relative contribution of antagonists of the various ionotropic glutamate receptors (NMDA, AMPA, KA) to the potential for treating migraine is under investigation. Preclinical data partially support the hypothesis that blocking any one of these ionotropic glutamate receptor subtypes is a potential approach.
LY293558 (Table 1) is a nonselective AMPA/KA (GluR5) receptor antagonist (Ki: GluR2 = 3.2 µ
LY293558 receptor profile

In vitro ligand binding affinity to various human receptors. GluR, Ionotropic AMPA (GluR1-4) and kainate (KA) (GluR5-7) type glutamate receptors. KA1,2, Ionotropic kainate type glutamate receptors 1 and 2. NMDA, N-methyl-D-aspartate. D, Dopamine receptor. 5-HT2, 5-Hydroxytryptamine-2 receptor. GABAA, Gamma-amino-butyric-acid type A receptor. H1, Histamine type 1 receptor.
IC50, concentration producing 50% of inhibition of inward currents activated by 10 µ
The preclinical profile of LY293558 raises the possibility that it could be effective in acute relief of migraine, without vasoconstrictive liabilities. Accordingly, we hypothesized that LY293558 is effective in clinical migraine and tested the hypothesis in a randomized controlled clinical trial. The primary objective of the study was to determine the efficacy of an intravenous (IV) infusion of LY293558 compared with placebo during a moderate to severe acute migraine attack. Secondary objectives were to assess the safety and tolerability of LY293558, and to compare the safety and tolerability of IV LY293558, SC sumatriptan 6 mg, and placebo.
Methods
Participants
Those patients eligible for participation in the study were male or female migraineurs in otherwise good health, at least 18 years old, who experienced episodes of migraine, with or without aura (International Headache Society categories 1.1 and 1.2) (15), for at least one year prior to presentation. They must have had at least one, but not more than 15, migraine episodes per month for each of the six months prior to the study. Pregnant or breastfeeding women were excluded. Women of childbearing potential were excluded initially but were subsequently admitted following negative embryo-fetal developmental preclinical toxicity tests. Every effort was made to enroll only patients who had the ability to understand and comply with instructions. An eligible attack for the study had to be a moderate or severe migraine with or without aura, with time from onset to administration of study drug ≤ 8 h. Also, the patient could not have taken any treatment for the attack prior to arrival at the clinic for study drug administration.
Study design
The study was a multicentre, randomized, triple-blinded (subject-, investigator-, sponsor-blinded), placebo-controlled, parallel-group trial of 6 mg SC sumatriptan, 1.2 mg/kg IV LY293558, or placebo. Five sites participated in the study. Randomization occurred at the site level. Allocation of patients was balanced between treatments with a block size equal to 3. Randomization code was kept under lock, and was only accessed by the pharmacist or his/her designee.
The dose of LY293558 was based on the maximally tolerated dose (MTD) as determined from a previous healthy volunteer study (13). The 6-mg SC sumatriptan is the clinically recommended and commercially available dose.
Screening procedures at the initial visit included a medical history, physical examination (including vital signs and measurements of body weight, height, and temperature), clinical laboratory tests (including pregnancy test for women who could not confirm their inability to bear children), electrocardiogram (ECG), and a urine drug screen.
After screening, each patient was instructed to contact the clinic by phone when he or she experienced a migraine attack. If eligible, the patient came to the clinic within 8 h of symptom onset. He/she was given a 3-digit identification number and was randomly assigned to receive one of the following treatments: a 1.2 mg/kg IV LY293558 infusion over 15 min, a 6 mg SC injection of sumatriptan, or placebo. A ‘double-dummy’ technique was used in this study, such that patients in all three groups received both an IV infusion and a SC injection. At 2 h postdosing, a rescue medication, excluding ergot derivatives, was available at the patient's request.
The study was conducted in accordance with ICH Guidelines for Good Clinical Practice. Regulatory approval was obtained at each investigative site. Also, each patient signed a voluntary informed consent to participate in the study.
Efficacy measures
Treatment efficacy was measured using the Migraine Headache Assessment Questionnaire© (MHAQ) that was completed predose (baseline) and at 15, 30, 45, 60 and 90 min and 2, 3, 4 and 24 h postdosing. The MHAQ assessed a number of variables, including date, time, intensity of the headache, and the presence of nausea, phonophobia, and photophobia. Patients completed the 24-h assessment at home, and reported to the investigator at the follow-up visit, which occurred within one business day of study drug administration.
The severity of headache and associated symptoms such as nausea, vomiting, photophobia, and phonophobia was scored on a 4-point scale(0 = no symptom, 1 = mild, 2 = moderate, 3 = severe). The primary efficacy variable was response at 2 h (percent of patients with headache improvement to mild or no pain at 2 h postdosing), and the primary treatment comparison was between LY293558 and placebo. Additional efficacy measures included ≥ 2-point improvement in pain score (percentage of patients with at least 2-point improvement in pain score from baseline), pain-free rate (percentage of patients with no headache at 2 h), sustained response rate (percentage of patients with response at 2 h and no rescue or recurrence from 2 to 24 h), sustained pain-free rate (percentage of patients pain-free at 2 h and no rescue or recurrence from 2 to 24 h), and recurrence rate (percentage of patients with 2-h response who experience a 2/3 headache from 2 to 24 h). Response rates, pain free rates, and ≥ 2 point improvement in pain score were also analysed at 15, 30, 45, 60, and 90 min postdosing. Non-pain efficacy measures were relief of nausea, photophobia, or phonophobia at 2 h (% of patients with no symptom at 2 h, from a 2/3 baseline score).
Adverse events
Adverse events (AEs) and safety parameters were recorded for 24 h. Safety data were monitored for treatment emergent signs and symptoms by a clinical research physician, and appropriate medical care was maintained throughout the study.
Sample size and statistical analyses
Assuming a 2-h response rate of 75% for active therapy vs. 23% for placebo, 15 evaluable patients per treatment group were sufficient to detect a significant treatment difference with at least an 80% power and a two-sided α-error of 0.05. Safety and efficacy data were combined for analysis from this multicentre study. Treatment differences for binary variables were assessed using the χ2 test. Also, Fisher's exact test was conducted posthoc in order to analyse the efficacy data. Continuous variables were analysed using a one-way (treatment) analysis of variance. Analyses were conducted on data obtained from evaluable patients, i.e. those who received study drug and had at least one assessment postdrug administration.
Results
Study group
A total of 100 patients were screened between August 1999 and September 2000 (Fig. 1). Forty-five patients were randomized but one person withdrew consent before receiving study drug. The total number of evaluable patients was 44, with n = 16 for placebo, n = 13 for LY293558, and n = 15 for sumatriptan. The mean age of the evaluable patients was 40 years and 55% were women. The mean time to treatment for all patients was 03.85 h. Ten (62.5%) patients from the placebo group, 9 (69.2%) from the LY293558 group and 11 (73.3%) from the sumatriptan group reported prior triptan use. Nine (56.3%) patients in the placebo group, 9 (69.2%) in the LY293558 group and 10 (66.7%) in the sumatriptan group reported severe pain at entry. One patient in the placebo group treated a mild headache. Baseline characteristics were comparable between groups (Table 2).

Flow of participants.
Baseline characteristics

One patient was randomized but withdrew consent before receiving study drug;
One patient treated a mild headache.
Efficacy outcomes
Response rates at 2 h after infusion were 25% (4/16) for placebo, 69% (9/13) for LY293558 (P = 0.017 vs. placebo), and 87% (13/15) for sumatriptan (P = 0.001 vs. placebo) (Fig. 2a). Significant differences from placebo were observed as early as 30 min postdosing for sumatriptan and 45 min for LY293558. Also, 54% of patients treated with LY293558 became pain-free at 2 h (P = 0.004 vs. placebo), compared with 6% of the placebo group and 60% of the sumatriptan group (P = 0.001) (Fig. 2b). Furthermore, a greater proportion of subjects receiving either LY293558(8/13–62%, P = 0.006) or sumatriptan (12/15–80%, P < 0.001) reported an improvement of 2 or more points at 2 h than those on placebo (2/13–13%). Both LY293558 and sumatriptan were superior to placebo (P < 0.01) on all other secondary pain efficacy outcomes (sustained response; sustained pain free; relief of associated symptoms). None of the LY293558-treated patients who responded to treatment, one who was on sumatriptan, and two of four who were on placebo complained of headache recurrence. Finally, 4 (31%) patients on LY293558, 14 (88%) on placebo, and 2 (13%) on sumatriptan used a rescue medication 2h poststudy drug administration (P = 0.002 LY293558 vs. placebo; P = 0.001 sumatriptan vs. placebo).
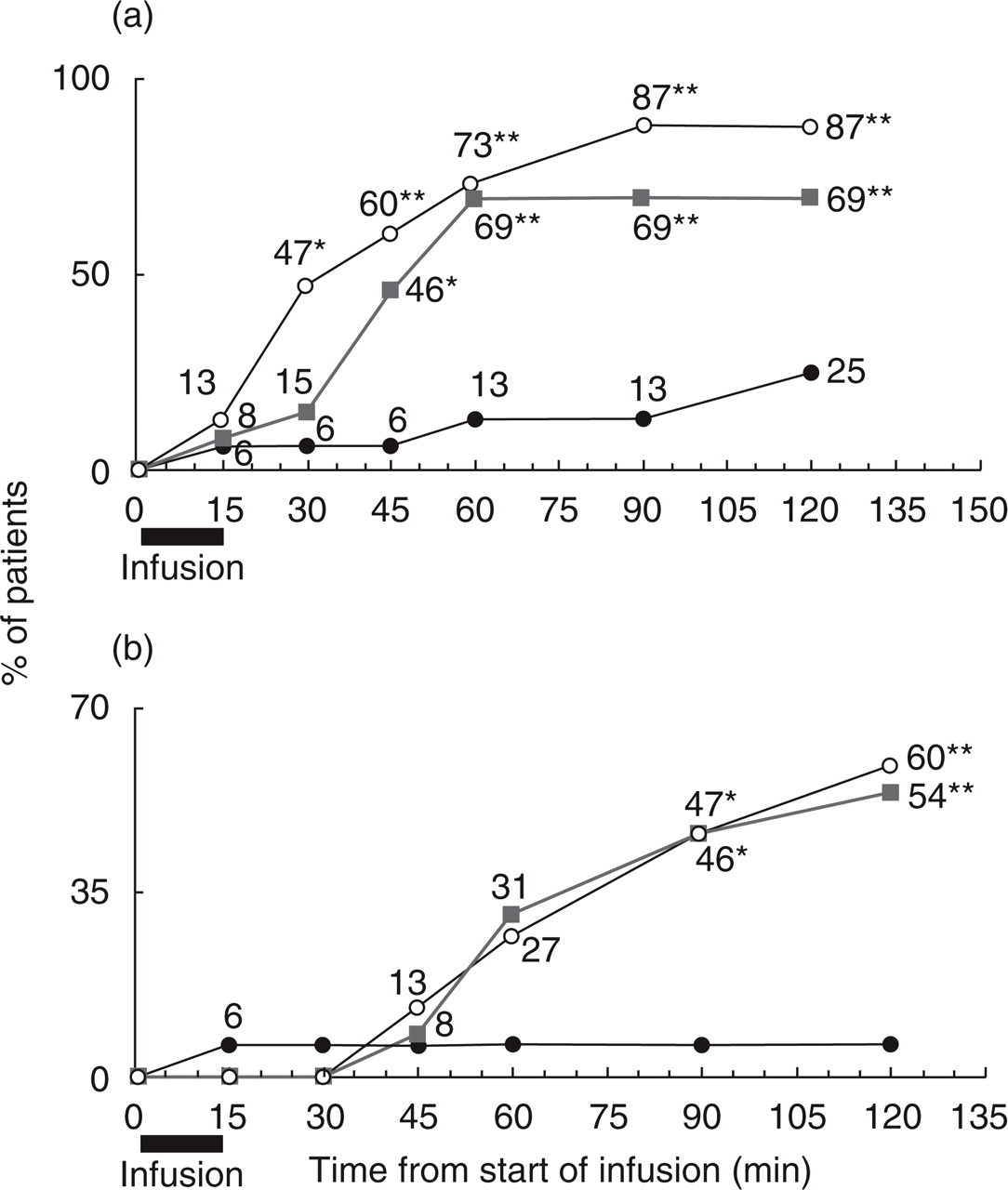
Pain efficacy outcomes. (a) response and (b) pain free rates in the first 2 h post-treatment. •= placebo, ▪= LY293558, ○= sumatriptan. ∗P < 0.05, ∗∗P < 0.01 (vs. placebo).
Post-hoc analysis
Efficacy data of patients on LY293558 were compared to those on placebo using Fisher's exact test. The 2-h response rate (P = 0.027), 2-h pain free rate (P = 0.01), 2 or more points improvement in headache score (P = 0.016), use of rescue medications (P = 0.03), and relief of associated symptoms (P < 0.01), all confirmed that LY293558 is more effective than placebo.
Safety
Adverse events occurred in 2/13 (15%) patients in the LY293558 treatment group, 5/16 (31%) patients in the placebo group, and 8/15 (53%) patients in the sumatriptan group (Table 3). In the group receiving LY293558, dizziness and sedation/drowsiness were the most common AEs. In the placebo group, sedation/drowsiness was the most common AE and dizziness was the second most common. Heaviness/tingling, sedation/drowsiness, and warmth were the most commonly reported AEs for the sumatriptan group. Two of 13 patients who received sumatriptan reported chest/throat symptoms.
Adverse events (AEs) reported by >10% of patients

Adverse events were moderate (n = 1) or severe (n = 1) in the LY293558 group, mild (n = 4) or severe (n = 1) in the placebo-treated patients, and mild (n = 7) or moderate (n = 1) in those who received sumatriptan.
Discussion
The results of this exploratory proof of concept study indicate that SC sumatriptan and IV LY293558 are both superior to placebo in the treatment of acute, moderate or severe migraine attacks. More than two-thirds of patients who received IV LY293558 reported a reduction of headache pain from moderate-to-severe to mild-or-no pain at 2 h, and more than half were pain-free at 2 h without headache recurrence or need for rescue therapy. Also, LY293558 was effective in relieving nausea, photophobia, and phonophobia, complaints that impart significant disability during migraine. Moreover, LY293558 was at least as well tolerated as placebo.
The significant sustained response (69%; placebo = 6%) and sustained pain-free (60%; placebo = 0%) rates, and the total absence of headache recurrence in patients on LY293558, were not predictable based on the short plasma half-life (approximately one hour) of IV LY293558, indicating a potential pharmacokinetic/pharmacodynamic mismatch. There are a few possible explanations for this observation. First, we may speculate that only a small amount of LY293558 needs to reach the target site with fast on- and slow off-receptor rate. This hypothesis is worthy of exploration. Second, LY293558 may induce beneficial secondary pharmacologic effects. The colocalization of glutamate receptors with 5HT1B/1D/1F receptors (16) and the release of CGRP by activation of glutamate receptors (17) may indicate that LY293558 suppresses the release of pro-inflammatory neuropeptides that perpetuate the migraine attack. The possibility of a metabolite-related effect is unlikely, since LY293558 is > 90% excreted unchanged in the urine (Data on file, Eli Lilly & Co., Indianapolis, IN, USA). Third, the observation of no recurrence on LY293558 could be a chance occurrence given the small sample size.
Not only did LY293558 demonstrate efficacy in all variables, but also the incidence of adverse events was low (15%). Especially promising was the lack of chest symptoms or cardiovascular signs with LY293558. These side-effects have contributed to limiting the use of triptans and ergots in several groups of migraineurs. The low incidence of AEs on IV LY293558 in this trial could be the result of using a dose slightly lower than that associated with relatively few symptoms in our healthy volunteer study (13). On the other hand, the low incidence of visual symptoms on LY293558 (8%) was unanticipated, given that the median MTD (mostly visual symptoms related) in the healthy volunteer study was 1.3 mg/kg (13). Migraineurs have demonstrated a poor threshold for contrast discrimination (18), and may be unable to identify minor visual disturbances. Also, migraineurs are photosensitive during an attack, and they may not spontaneously report additional minor changes in vision.
In conclusion, we have demonstrated that LY293558 is effective in the treatment of acute migraine, and that AMPA/GluR5 antagonism may provide a novel target for migraine therapy that circumvents the vasoconstrictive liability of 5-HT1B agonism, a property shared by triptans and ergots. In the absence of vasoconstrictive effects, this potential therapy is quite attractive as it could be extended to patients with coronary artery risk factors, a group in whom current migraine-specific therapies are contraindicated. This hypothesis needs further testing before making firm conclusions. The results of this randomized trial are promising, but this was only a pilot study of LY293558 in migraine with a relatively small number of patients. Further evaluation with larger patient groups is needed to better quantify the efficacy and assess the safety of LY293558 and other ionotropic glutamate receptor antagonists in acute migraine.
Financial disclosures and contributions to the study
NM Ramadan (corresponding author): Employee of Eli Lilly & Co. at the time of manuscript initial submission. Contributed to the design and conduct of the study, analysis of results, manuscript preparation and coordination.
CN Sang: Consultant and research grant recipient, Eli Lilly & Co. Contributed to the design and conduct of the study, analysis of results, and manuscript preparation. Advisory panel and/or research grant support recipient and/or served as a consultant for Endo Pharmaceuticals, Forest Laboratories, Johnson & Johnson (Janssen Pharmaceuticals and Ortho-McNeil), Merck & Co., Pfizer, Schwarz Pharma Inc., Syntem Inc., and UCB Pharma.
RG Wallihan: Employee of Eli Lilly & Co. at the time of manuscript writing. Contributed to writing of manuscript.
AS Chappell: Employee of Eli Lilly & Co. Contributed to the design of the study.
FG Freitag: Conducted research for and/or served on the speakers’ bureau and/or served as a consultant for Abbott Laboratories, Allergan, Bayer Pharmaceuticals, Bristol-Myers-Squibb, Elan Pharmaceuticals, GlaxoSmithKline, Merck & Co., Novartis, Pfizer, Pozen, Winston, AstraZeneca, CAPNIA, Pharmacia, Epicept, Janssen Pharmaceuticals, and Wyeth Ayerst Laboratories. Contributed to the study as primary investigator on the trial and manuscript preparation.
TR Smith: Consultant for Eli Lilly & Co. and research grants/support recipient from Eli Lilly & Co. Contributed to the design and implementation of the study. Served on the advisory committee that evaluated the data and planned the publication process. Reviewed the manuscript.
SD Silberstein: Advisory panel and/or speakers’ bureau for Abbott, Allergan, AstraZeneca, Elan Pharmaceuticals, Eli Lilly & Co., Johnson & Johnson (Ortho-McNeil), Merck, GlaxoSmithKline. Research support from Allergan, Eli Lilly & Co., GlaxoSmithKline, Janssen, Johnson & Johnson, Merck, Ortho-McNeil, Pfizer, Robert Wood Johnson, UCB Pharma, Vernalis. Unrestricted educational grants from Abbott, Allergan, AstraZeneca, Bristol-Myers-Squibb, Eli Lilly & Co., GlaxoSmithKline, Johnson & Johnson, Merck, Parke Davis. Contributed to the design of trial, enrolment of patients, manuscript preparation and review.
KW Johnson: Employee of Eli Lilly & Co. Contributed to the preclinical discovery of LY293558 as a potential migraine therapy, and manuscript preparation.
LA Phebus: Employee of Eli Lilly & Co. Generated preclinical data on the compound.
D Bleakman: Employee of Eli Lilly & Co. Pharmacologist involved in in vitro discovery of glutamatergic activity of LY293558 and its experimental development.
PL Ornstein: Employee of Eli Lilly & Co. Conceived of and is the key inventor for LY293558. Designed and implemented the synthesis of the compound.
MB Arnold: Employee of Eli Lilly & Co. Contributed to the synthesis of LY293558.
SJ Tepper: Advisor to Eli Lilly & Co., and GlaxoSmithKline; Research grant from Eli Lilly & Co. and GlaxoSmithKline; Speakers’ Bureau, GlaxoSmithKline. Met with other authors in preparation of the study, participated in the study, implemented the study, helped edit and approved the manuscript.
F Vandenhende: Employee of Eli Lilly & Co. Contributed to statistical analyses and manuscript preparation.
Footnotes
Acknowledgements
The authors wish to thank the following people for their contribution to the success of the trial: J Bjerke, J Burgess, M Gates, K Hoo, WW Offen and A Ogden (Eli Lilly & Co.); J Berg, L Dobosh, L Jinga and A O’Donnell (Massachusetts General Hospital); Allelix Biopharmaceuticals, Inc.; S Diamond, M. Diamond and G Urban (Diamond Headache Clinic); M Holden, S Rocco and D Sullentrup (Mercy Health Research); F Arrowsmith, I Gamerman, AM Rapoport and F Sheftell (New England Center for Headache). The study was funded by Eli Lilly & Co.