
Abstract
Background: Cortical spreading depression is the electrophysiological substrate of migraine aura, and may trigger headache. Recently, chronic treatment with five migraine prophylactic drugs was shown to suppress cortical spreading depression, implicating spreading depression as a common therapeutic target in migraine prophylaxis.
Materials and methods: In order to assess the negative predictive value of spreading depression susceptibility as a preclinical drug screening tool, we tested oxcarbazepine, an anti-epileptic ineffective in migraine prophylaxis. Valproate served as the positive control. Cortical spreading depression susceptibility was measured in rats using topical KCl or electrical stimulation.
Results: Oxcarbazepine did not suppress spreading depression either after a single dose or after daily treatment for 5 weeks. As previously shown, valproate suppressed spreading depression susceptibility after chronic dosing, while a single dose was ineffective.
Conclusions: These data provide further support for spreading depression as a relevant target in migraine prophylaxis, and demonstrate the predictive utility of employed spreading depression models.
Introduction
Migraine is a chronic, often disabling, neurological condition with a 1-year prevalence of approximately 18% in women and 6% among men in the US. Prophylaxis is recommended when patients experience more than 3–5 attacks per month. Unlike targeted abortive and analgesic treatments, prophylactic drugs have largely been discovered serendipitously (e.g. propranolol) or based on presumed class effects (e.g., anti-epileptics) (1). Although some anti-epileptic drugs have been efficacious in migraine prophylaxis, others have been ineffective (e.g., oxcarbazepine) (2). Recently, two anti-epileptic drugs (i.e. valproate, topiramate) and three other unrelated migraine prophylactic drugs (i.e. propranolol, amitriptyline, and methysergide) were all found to suppress cortical spreading depression (CSD) (3,4), an electrophysiological phenomenon widely accepted as the substrate of migraine aura and possibly a trigger for headache. Importantly, all five drugs required chronic treatment for several weeks to achieve CSD suppression in one study (3), reminiscent of the gradual build up of their clinical efficacy in migraine. These results suggested that CSD may be a common pathophysiological target for some established or investigational migraine prophylactic drugs.
Experimental models of CSD as a surrogate for aura can be useful in preclinical drug screening for migraine prophylaxis. Unlike their positive predictive value, the negative predictive value of experimental CSD susceptibility models has not been tested, because only a few drugs have been rigorously studied and found ineffective in clinical trials of migraine prophylaxis (1,3–5). Recently, oxcarbazepine was conclusively ineffective as a migraine prophylactic drug in a randomized, double-blind, placebo-controlled clinical trial (2). Here, we report that oxcarbazepine does not suppress CSD susceptibility in established experimental models in vivo, providing further evidence for the predictive utility of CSD susceptibility as a preclinical drug screening tool in migraine prophylaxis.
Materials and methods
Drugs and experimental groups
Systemic physiological parameters
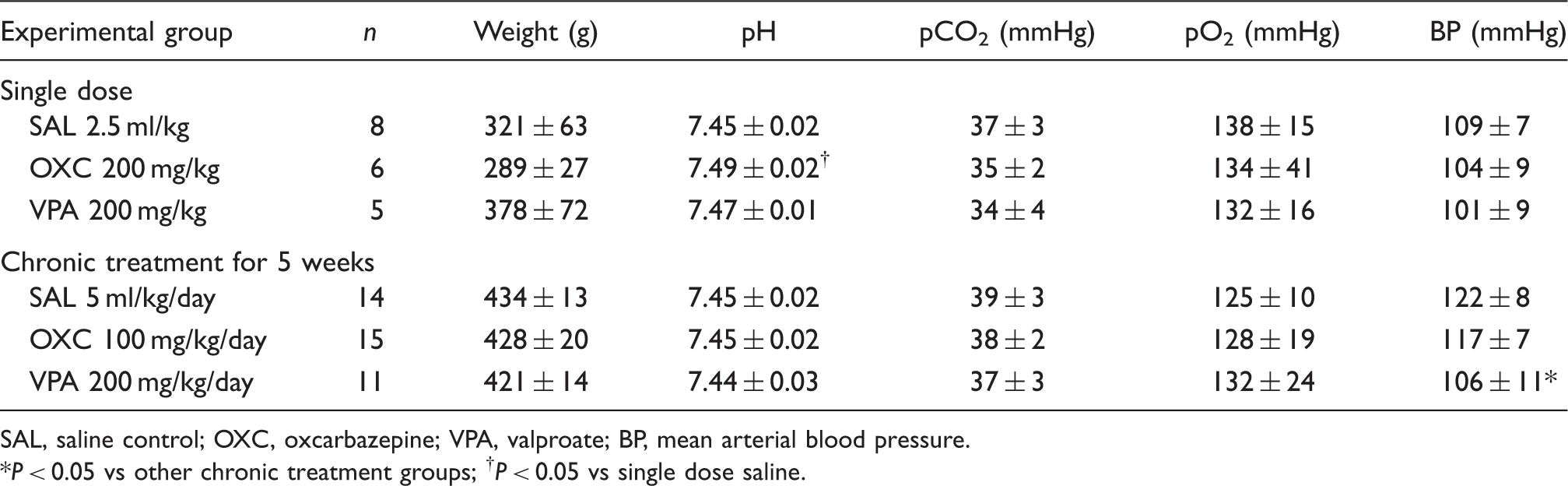
SAL, saline control; OXC, oxcarbazepine; VPA, valproate; BP, mean arterial blood pressure.
*P < 0.05 vs other chronic treatment groups; † P < 0.05 vs single dose saline.
Procedures
Institutional guidelines for animal care and use for research purposes were strictly followed, and study protocol was approved by institutional review board. Rats were anaesthetized (isoflurane 5% induction, 1% maintenance, in 70% N2O/30% O2), paralyzed (pancuronium 0.4 mg/kg), and intubated via a tracheostomy for mechanical ventilation (SAR-830; CWE, Ardmore, PA, USA). Arterial blood gases and pH were measured every 30 min and ventilation adjusted to maintain arterial pCO2 between 35–45 mmHg (Corning 178; Corning, NY, USA). Continuous measurement of blood pressure (PowerLab; ADInstruments, Colorado Springs, MO, USA) and blood sampling were performed via a femoral artery catheter. Rectal temperature was kept at 37.0 ± 0.1°C using a thermostatic heating pad (FHC, Bowdoinham, ME, USA). Level of anaesthesia was maintained throughout the experiment to eliminate cardiovascular response to tail pinch. In all treatment groups, systemic physiological parameters were within normal range (Table 1).
Rats were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA) and burr holes were drilled bilaterally under saline cooling at (mm from bregma): (i) posterior 7.0, lateral 2.0 (occipital, 1 mm diameter for KCl application or electrical stimulation); (ii) posterior 5.0, lateral 2.0 (frontoparietal, 0.5 mm diameter for electrode 1); (iii) posterior 3.0, lateral 2.0 (frontal, 0.5 mm diameter for electrode 2). Of note, these recording co-ordinates differed from our previous study, and were closer to the KCl application site (3). Dura overlying the occipital cortex was gently removed and care was taken to avoid bleeding. The steady (DC) potential and electrocorticogram were recorded with glass micropipettes filled with 200 mM NaCl, 300 µm below pia (Axoprobe-1A; Axon Instruments, Burlingame, CA, USA). Ag/AgCl reference electrode was placed subcutaneously in the neck. After surgical preparation, cortex was allowed to recover for 15 min under saline irrigation. The data were continuously recorded using a data acquisition system for off-line analysis (ADInstruments).
Cortical spreading depression susceptibility
We assessed CSD susceptibility using two independent methods, KCl or electrical stimulation, as described previously (3). For KCl-induced CSD susceptibility, we placed a cotton ball (1.5 mm diameter) soaked with 1 M KCl on the pial surface and kept it moist by placing 5 µl of the same KCl solution every 15 min. The total number of KCl-induced CSDs detected at either recording site during 60 min KCl application was counted. In addition, we determined the incidence of propagation failure between the two recording sites in all treatment groups, and expressed this as the number of CSDs failed to appear at electrode 2 as percentage of total CSDs recorded at electrode 1. After the end of KCl stimulation, electrical threshold for CSD was determined in the opposite hemisphere by direct cortical stimulation using a stimulator (Grass Instruments, West Warwick, RI, USA), a constant current unit (WPI, Sarasota, FL, USA), and a bipolar stimulation electrode placed on the pial surface (400 µm tip diameter, 1 mm tip separation; FHC). Cathodal square pulses of increasing intensity (100–4000 microcoulomb, µC) were applied at 5-min intervals by adjusting the current and duration of stimulus until a CSD was observed. At 1 mA current, pulses of 100, 200, 300, and 400 ms were applied, followed by 2 mA current of 300, 400, and 500 ms. If CSD did not occur, additional stimuli of 3 mA, 400 ms, and 4 mA, 400, 500, 1000 ms were applied. The logarithmic stepwise escalating stimulation protocol was chosen for its ability to distinguish group differences in CSD threshold based on our prior experience (3). As previously observed, electrical stimulation threshold for CSD showed more variability compared to the frequency of CSDs evoked by topical KCl, which is often attributed to variations in the stimulus current-density geometry between the electrode and the cortex. In addition, we calculated CSD propagation speed by dividing the distance (mm) between the two recording electrodes by the CSD latency (min) between these sites. CSD amplitude and duration at half-maximal amplitude were also measured.
Statistical analysis
The systemic and electrophysiological data and the number of CSDs after topical KCl were compared using one-way analysis of variance followed by Dunnett’s multiple comparisons versus control. Electrical stimulation threshold was analyzed using Kruskal–Wallis one-way analysis of variance on ranks, followed by Dunn’s multiple comparisons versus control. Data were expressed as mean ± SD for systemic physiology, electrophysiology, and number of KCl-induced CSDs, or as median (25–75% range) for electrical CSD threshold.
Results
Oxcarbazepine either as a single dose (200 mg/kg, p.o.) administered 90 min before CSD testing or chronically (50 mg/kg, p.o., twice a day) for 5 weeks did not suppress CSD susceptibility as measured by the frequency of KCl-induced CSDs or the electrical stimulation threshold for CSD induction (Figures 1 and 2; Table 2). As reported previously, a single dose (200 mg/kg, i.v.) of valproate administered 60 min before KCl application also failed to suppress KCl-induced CSDs or elevate the electrical threshold (3). In contrast, chronic treatment (200 mg/kg, i.p., once a day for 5 weeks) did reduce the frequency of KCl-induced CSDs and elevate the CSD threshold.
CSD susceptibility assessed by topical KCl or electrical stimulation after 5 week saline, oxcarbazepine or valproate treatment. Left panel: Representative extracellular micro-electrode recordings of repetitive CSDs triggered during 60 min topical KCl application (1 M). Valproate (VAL, 200 mg/kg/day, i.p. for 5 weeks) but not oxcarbazepine (OXC, 100 mg/kg/day, p.o. for 5 weeks) decreased the number of CSDs triggered by KCl compared to saline controls (SAL). Right panel: Representative tracings showing direct cortical electrical stimulation using cathodal square pulses of escalating intensities and alternating polarity until a CSD is triggered. Chronic valproate but not oxcarbazepine elevated the electrical threshold for CSD induction. Stimulus artefacts (vertical deflections) were trimmed for clarity. CSD susceptibility assessed by topical KCl or electrical stimulation after a single dose or chronic treatment with oxcarbazepine or valproate. Oxcarbazepine did not reduce the number of CSDs triggered during 1 h topical KCl (1 M) application (left upper and lower graphs) or elevate the cathodal stimulation threshold for CSD induction (right upper and lower graphs), either after a single dose (200 mg/kg, p.o.; upper left and right graphs) or 5 weeks of chronic daily dosing (100 mg/kg/day; lower left and right graphs). A single dose of valproate (200 mg/kg, i.v.) was similarly ineffective on these CSD susceptibility measures, where as chronic treatment for 5 weeks (200 mg/kg/day, i.p.) both reduced the number of KCl-induced CSDs and elevated the electrical threshold. Each data point represents one animal. Note the logarithmic scale for CSD threshold data (right upper and lower graphs). Group means and standard deviations for CSD frequency, and medians and interquartile ranges for electrical threshold are provided in Table 2. SAL; saline controls; OXC, oxcarbazepine; VPA, valproate. *P < 0.05 versus other groups. Electrophysiological measures of CSD SAL, saline control; OXC, oxcarbazepine; VPA, valproate; Amp, DC shift amplitude; Dur, DC shift duration at half amplitude. E1 and E2, electrodes 1 (proximal) and 2 (distal; see Materials and methods). *P < 0.05 vs other chronic treatment groups; †
P < 0.05 vs single dose or chronic saline.

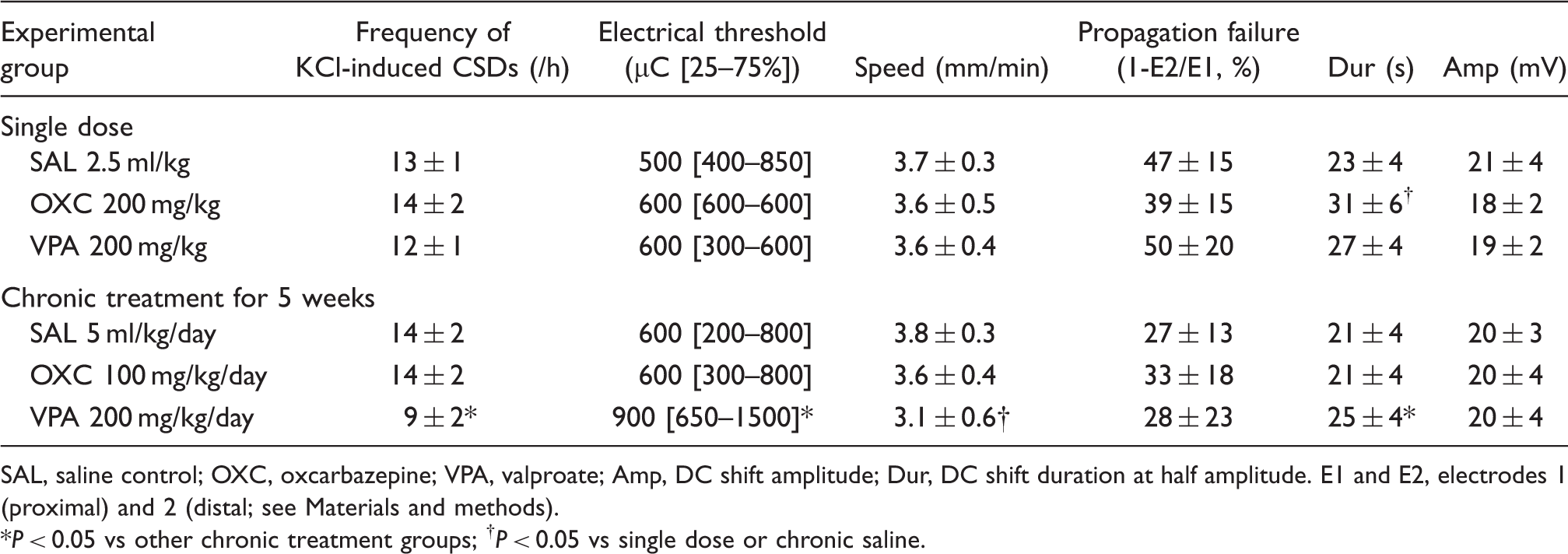
The CSD propagation speed was reduced in chronic valproate group only (Table 2). The amplitude and duration of CSDs were not altered in any treatment group, except for mildly prolonged CSD durations after chronic valproate and single dose oxcarbazepine treatments compared to saline. Neither drug significantly altered the incidence of propagation failure between the proximal and distal recording sites during KCl-induced repetitive CSDs.
Discussion
The mechanistic basis of migraine prophylaxis remains elusive. Recent data implicated CSD suppression as a final common mechanism shared by five seemingly unrelated prophylactic drugs (3). Here, we provide evidence suggesting that oxcarbazepine, an anti-epileptic clinically ineffective in migraine (2), does not suppress CSD susceptibility even after chronic treatment. Together, these data provide further support for the positive and negative predictive value of CSD susceptibility models for drug efficacy in migraine prophylaxis (1).
The lack of efficacy of oxcarbazepine on CSD either acutely (200 mg/kg) or after chronic treatment (100 mg/kg/day) is consistent with its lack of efficacy in migraine prophylaxis at doses as high as 20 mg/kg/day in humans (2). Upon oral administration, oxcarbazepine is rapidly absorbed with excellent bioavailability, and metabolized into its biologically active monohydroxy derivative which readily crosses the blood–brain barrier. Peak plasma and brain tissue levels are reached 1 h after an oral dose in rats, with a plasma half-life longer than 4 h (9,10). Therefore, our timing of CSD testing starting 90 min after a single oxcarbazepine dose corresponded well with the anticipated plasma levels. In additional experiments, we also waited for 180 min after oxcarbazepine administration and once again did not see CSD suppression (n = 3, data not shown). The lack of efficacy was not due to insufficient dosing because, in experimental models of epilepsy and pain, oxcarbazepine shows efficacy at lower doses than used in our study (11). Indeed, in most studies the highest dose tested was 100 mg/kg, and mild toxicity started at chronic doses exceeding 200 mg/kg/day without losing efficacy (12–14).
To date, many migraine prophylactic drugs have been tested on CSD (1). However, the experimental medium (i.e. in vitro vs in vivo) and species, drug doses, treatment routes and durations, methods used to trigger and detect CSD, and measured end-points have been highly variable, making it difficult to reach a consensus on whether a given drug inhibits CSD. Similarly, many drugs have been clinically tried in migraine prophylaxis, most showing some efficacy but few achieving rigorous clinical trial standards. Therefore, available data are insufficient to assess the predictive power of experimental CSD susceptibility models, and testing more drugs with conclusive clinical trial evidence will be important to improve our confidence in these models.
Conclusions
Valproate but not oxcarbazepine suppress CSD susceptibility, consistent with the reported clinical efficacies of these drugs in migraine prophylaxis. These data support the negative predictive value of the experimental CSD susceptibility models, arguably more important than the positive predictive value for their overall utility as preclinical drug screening tools in migraine prophylaxis.
Footnotes
Acknowledgements
This work was supported by the National Institute of Health (NS061505, CA) and Deutsche Forschungsgemeinschaft (HO 4211/1-1, UH).