
Abstract
Renal chloride metabolism is currently poorly understood but may serve as both a diagnostic and a treatment approach for acute kidney injury. We investigated whether plasma chloride, ammonia and glutamine as well as urinary chloride, ammonium and glutamine concentrations may serve as markers for acute kidney injury in paediatric patients. We conducted a prospective observational trial in a tertiary care paediatric intensive care unit. Ninety-one patients after cardiopulmonary bypass surgery were enrolled. Plasma glutamine, creatinine, (serum) albumin, urinary electrolytes and glutamine were collected pre-cardiopulmonary bypass surgery, at paediatric intensive care unit admission, and at 6, 12, 24, 48 and 72 h after paediatric intensive care unit admission. The urinary strong ion difference was calculated. The median urinary chloride excretion decreased from 51 mmol/L pre-cardiopulmonary bypass to 25 mmol/L at paediatric intensive care unit admission, and increased from 24 h onwards. Patients with acute kidney injury had lower urinary chloride excretion than those without. The median urinary strong ion difference was 59 mmol/L pre-cardiopulmonary bypass, rose to 131 mmol/L at 24 h and fell to 20 mmol/L at 72 h. The plasma chloride rose from 105 mmol/L pre-cardiopulmonary bypass to a maximum of 109 mmol/L at 24 h. At 24 h the plasma chloride concentration was associated with the presence of acute kidney injury. There was no association between plasma or urinary amino acids and chloride excretion or kidney injury. In conclusion, renal chloride excretion decreased in all patients, although this decrease was more pronounced in patients with acute kidney injury. Our findings may reflect a response of the kidneys to critical illness, and acute kidney injury may make these changes more pronounced. Targeting chloride metabolism may offer treatment approaches to acute kidney injury.
Introduction
There are numerous studies that have focused on the association between acute kidney injury (AKI), intravenous electrolyte administration and critical illness outcomes.1 –6 It remains unclear, and results are divergent, whether the amount of intravenous chloride administration is or is not associated with morbidity and mortality in critically ill patients.3 –10 Similarly, some studies suggest that increased plasma chloride is associated with AKI.3 –10 An underlying assumption of all these studies is that hyperchloraemia results from exogenous chloride administration rather than a defect in chloride excretion. However, hyperchloraemia may result from high chloride administration, reduced renal chloride excretion, or a combination of both. Further, hyperchloraemia may be a consequence of or a contributor to AKI. However, none of the above cited studies investigated whether increased plasma chloride concentration may be the result of decreased chloride excretion. The divergent results regarding morbidity and mortality may stem from the fact that renal chloride excretion may determine the plasma chloride concentration far more than exogenous administration, and consequently any observed morbidity and mortality outcomes may be linked to renal chloride excretion more than intravenous chloride administration. Consequently, should altered renal chloride excretion be found in critical illness and AKI, targeting the renal chloride excretion pathway may offer a treatment approach to AKI, which so far is lacking.
One important pathway of chloride excretion is through the binding with ammonium, which is produced as ammonia in the proximal tubular cells as well as the intercalated cells of the collecting duct (ICCD). 11 Through binding chloride, ammonium aids in net acid excretion as well as nitrogen waste. 12 Renal tubular acidosis can result from defective ammonium production and/or excretion in the distal tubule, and is one of the main causes for hyperchloraemic metabolic acidosis.13 –15 Changes in chloride excretion should therefore be mirrored in ammonia excretion. Glutamine, as an ammonia donor, may be linked with AKI and renal electrolyte metabolism.
Cardiopulmonary bypass (CPB) surgery induces a post-CPB AKI rate of 5–60%,6,16 –19 which is why this cohort may lend itself to investigation of the association between renal injury and electrolyte findings. In an earlier study, a weak association was found between reduced urinary chloride concentration and AKI post CPB surgery. 20 Given the predictable critical illness state that children post CPB surgery experience, and earlier evidence of chloride metabolism changes after CPB surgery, we studied renal electrolyte metabolism patterns in this population, theorising that the post-CPB critical illness state and AKI may reflect any critical illness state more generally.
Methods and materials
Ethics approval
Ethics approval was obtained from the Children’s Health Queensland Human Research Ethics Committee (HREC/17/QRCH/310).
Study design
This was a non-randomised observational cohort study. It was a single-centre study carried out at a tertiary paediatric intensive care unit (PICU) from December 2017 to July 2020.
Participants
Patients 2 years of age or younger who underwent CPB surgery for a congenital heart defect were enrolled. The age cutoff was chosen given that the CPB-related critical illness state and AKI is most pronounced in this group. 21 Enrolment occurred consecutively as patients met the following eligibility criteria: presence of an intra-arterial line and indwelling urinary catheter (IUC), CPB surgery performed, and informed consent given. An age cutoff of less than or equal to 2 years of age was chosen given the effects of the critical illness state seen after CPB affect this group more readily, likely due to differing inflammatory response, high metabolic demand and a lower patient to circuit blood volume ratio. 21
The exclusion criterium was anuric AKI before CPB. Measurements were discontinued as soon as the IUC or intra-arterial line was removed for clinical indications. This was necessary given urinary ammonium measurements would become unreliable in a non-catheter, not freshly collected urine specimen. Consent was obtained either before enrolment or as a consent-to-continue. Prophylactic antibiotic treatment with cephazolin was administered before and for 48 h after CPB surgery as a standard therapy.
Study procedures
Urine collection was standardised. The IUC was clamped until 1–2 ml of fresh urine had accumulated, which was then aspirated and sent for analysis. Furosemide was the only diuretic administered to any of the study patients. Intravenous fluid management and the type of fluid prescribed was at the discretion of bedside clinicians.
Data collection and variables
At the time of enrolment patient demographics were collected. Paired urine and plasma electrolytes, glutamine and ammonia measurements were performed pre-CPB surgery (at induction of anaesthesia), at admission to the PICU, and at 6, 12, 24, 48 and 72 h after PICU admission. Plasma urea and creatinine were measured at each time point. Electrolyte content of intravenously administered fluid, fluid output, diuretic use as well as haemodynamic support were recorded starting pre-surgery. Included in these measurements was any fluid administered during surgery (including cardioplegia (Buckberg solution – see Supplementary Table 3)) as well as any blood products administered. 22 The cardiopulmonary bypass circuit was primed with packed red cells and Buckberg solution in a ratio of 4:1, as well as 20% albumin (100 mL) and Plasmalyte 148 solution. 23 When modified ultrafiltration was performed, the ultrafiltrate was analysed and the amount as well as the electrolyte content were recorded. The total electrolyte administration during surgery and the intensive care unit stay was calculated. Blood product electrolyte content was analysed and recorded.
We calculated the plasma strong ion difference (SIDPlasma) as described before.24,25
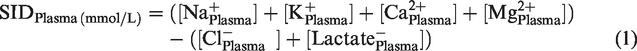
We calculated the urinary strong ion difference SID (SIDUrine) as described before.
26
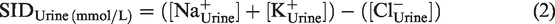
We calculated the urinary fractional excretion of sodium and chloride (FeNa and FeCl, respectively, with ‘X’ being either) as described before.
27
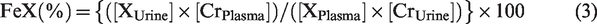
The ‘chloride balance’ was calculated by subtracting the total amount of chloride output from the total amount of chloride administered.
The ‘Kidney disease: improving global outcomes’ (KDIGO) criteria were used to classify kidney injury. 28 At each time point the degree of AKI was recorded, and its association with the measured plasma and urinary electrolytes/ammonium/glutamine concentrations was assessed. Diuretic use was recorded as having occurred in the time between the X and Y time points, and recorded as Yes/No for the Y time point.
Statistical analysis
Medians and interquartile ranges (IQRs) were used to describe most of the parameters of interest. To compare parameters between patients with no or AKI stage I, and patients with AKI stages II and III, Mann–Whitney U tests were used. Analyses were undertaken in StataSE 16.0 (Stata Corp Pty Ltd., College Station, TX, USA). Given this was an explorative study, we planned for a pragmatic, convenient sample size of 100 patients. Despite some differences in clinical characteristics at baseline, multivariable analyses were not undertaken due to the small sample size. Comparisons were considered significant at the P = 0.05 level, although all comparisons are acknowledged as exploratory; no adjustment for multiple comparisons was made.
Results
Of 129 screened patients, 14 were ineligible, eight were not approached for consent (due to rapidly deteriorating clinical situation, guardians not available for consent, or guardians distressed/overwhelmed), 10 declined consent and five did not receive CPB, leaving 91 participants in the cohort for analysis. For all participants, a complete dataset could be collected. Patient descriptors are shown in Table 1.
Patient demographics.
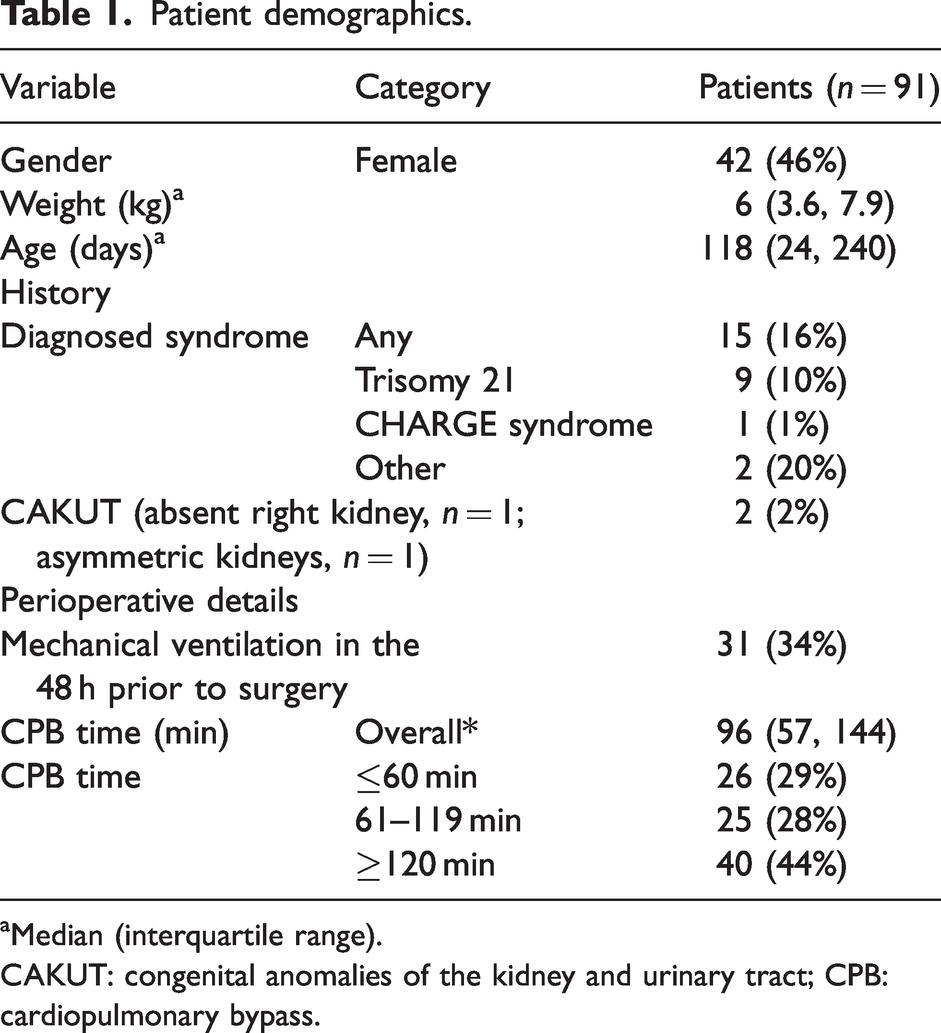
Median (interquartile range).
CAKUT: congenital anomalies of the kidney and urinary tract; CPB: cardiopulmonary bypass.
There was a marked initial decrease in urinary chloride excretion over the first 24 h (concurrent to an increase in plasma chloride), with a subsequent increase of urinary chloride excretion that started at and lasted beyond 24 h, particularly when compared with preoperative values (Figure 1 and Supplementary Table 1). This urinary chloride increase was evident independent of diuretic administration, although the increase in urinary chloride excretion from 24 h onwards was more pronounced in those patients who received diuretics (Supplementary Table 2). There was no difference in urinary chloride excretion between patients with or without AKI up to 12 h. At the 24-h time point patients with AKI had lower urinary chloride excretion than patients without AKI. No patient received furosemide in the operating theatre, or in the 12 h post PICU admission. Patients with AKI at any time point had a lower chloride excretion, and a slower recovery of the chloride excretion than seen in non-AKI patients (Figure 2). The fractional excretion of chloride decreased from 0.45% to 0.12% at 12 h and then increased to 3% at 72 h.
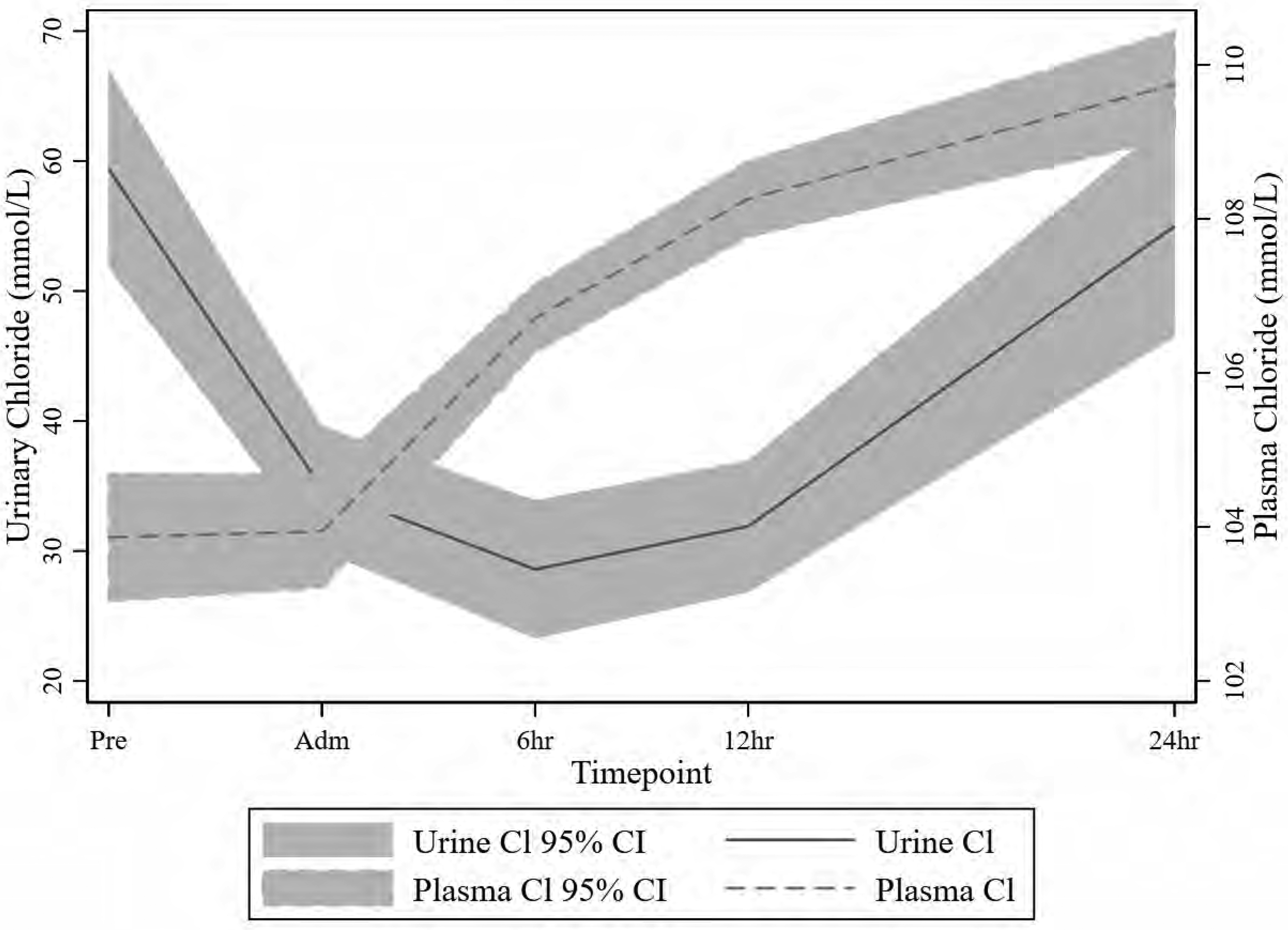
Fractional polynomial fit (with 95% confidence interval) of urinary and plasma chloride concentration over the time points.
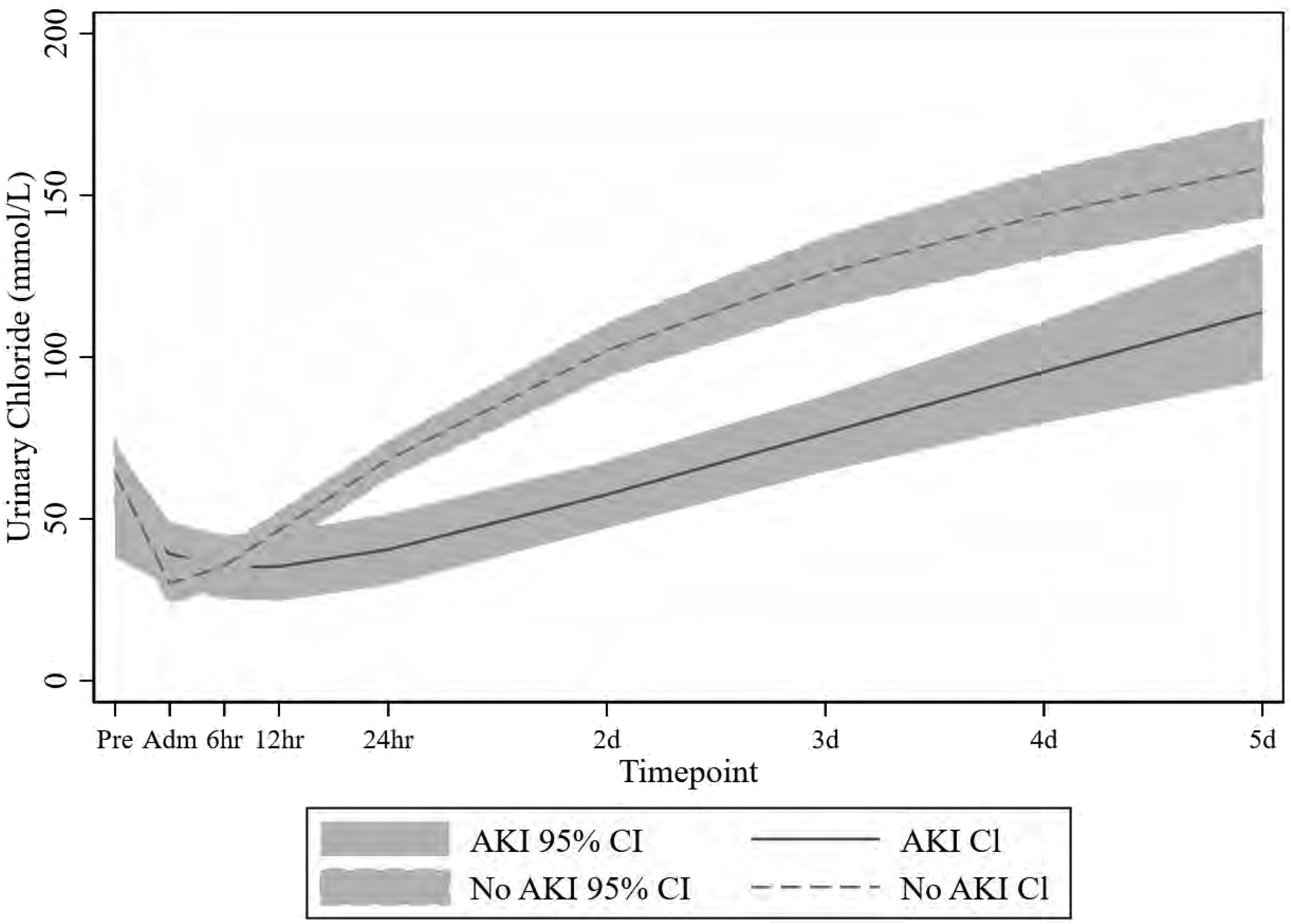
Fractional polynomial fit (with 95% confidence interval) of the urinary chloride excretion between acute kidney injury and non-acute kidney injury patients over the study time points.
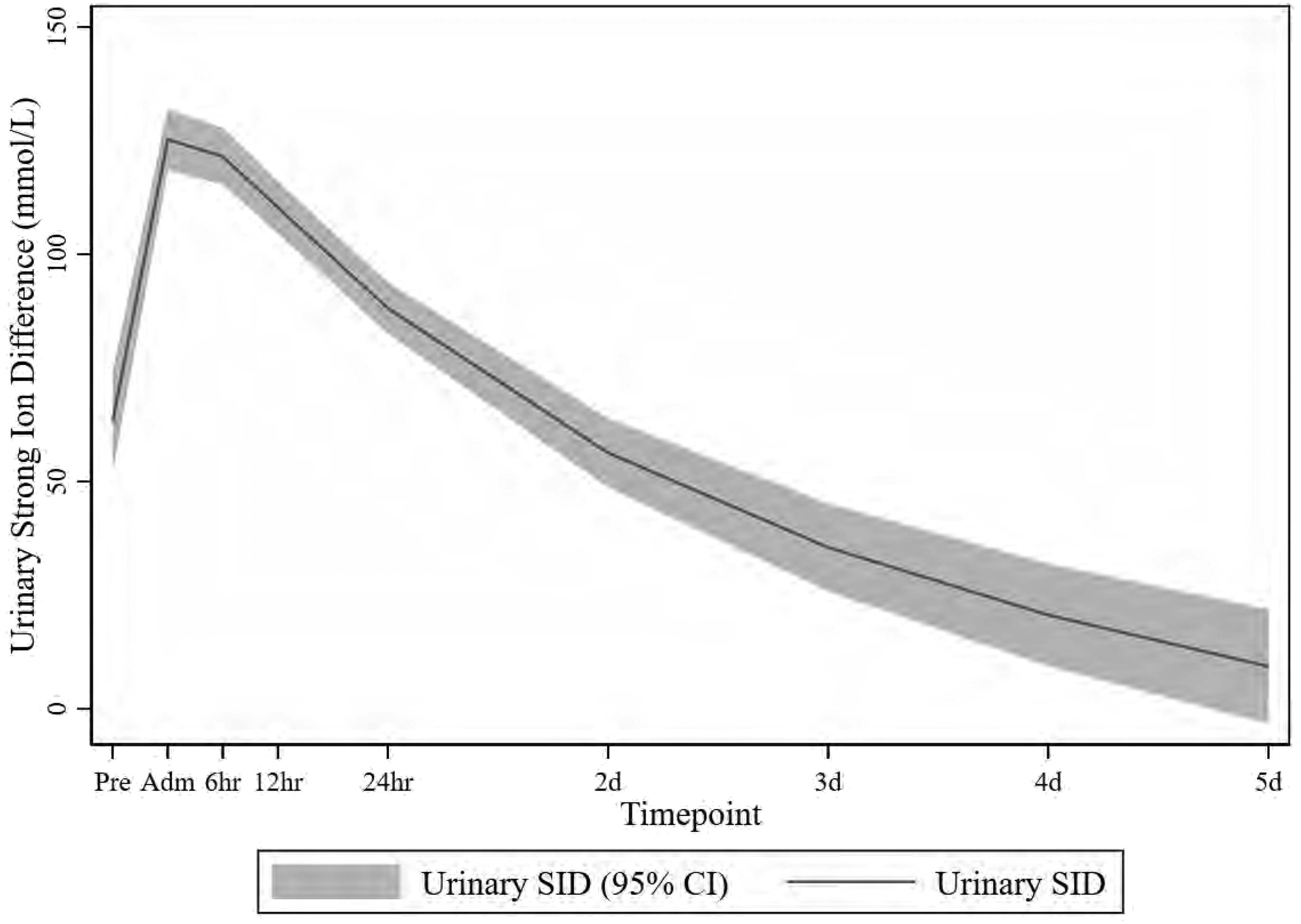
Fractional polynomial fit (with 95% confidence interval) of the urinary strong ion difference over the study time points.
The median plasma chloride was 105 (IQR 100, 107) mmol/L preoperatively and increased to a maximum of 110.5 (IQR 108, 114) mmol/L at 72 h in those not receiving diuretics (Supplementary Table 1). For the whole patient group there was a change in plasma chloride during the study (P <0.001 linear, quadratic and cubic terms) with a steady increase in plasma chloride concentration over the study period (Supplementary Table 1). The plasma chloride balance was positive at all time points after admission to surgery, but particularly so at 6 and 12 h.
There was no significant change in plasma sodium over the study. Urinary sodium (median (IQR)) rose from 30 (19, 62; n = 77) mmol/L preoperatively to 61 (35, 91; n = 85) mmol/L on admission and then to 31 (16, 64; n = 64) mmol/L during the first 24 h, and kept increasing thereafter. Median FeNa stayed less than 1.5% for the study until the 72-h time point (Supplementary Table 1).
The plasma SID was 36.8 mmol/L preoperatively, increased to 42.3 mmol/L at admission to the PICU and then decreased to 39.2 mmol/L and 38.2 mmol/L at 6 and 12 h, respectively. It then remained around 37 mmol/L for the remainder of the time points. The maximum plasma SID did not correlate with the lowest urinary chloride concentration.
The urinary ammonium (median (IQR)) decreased from 15 (7, 29) mmol/L preoperatively to 8 (4, 13) mmol/L at PICU admission but did not show a correlation with chloride thereafter (Figure 1 and Supplementary Table 1). Urinary ammonium concentrations were associated with urinary chloride concentrations only for the first 12 h. There was no difference in the urinary ammonium concentration between patients with or without AKI.
The preoperative urinary SID (median (IQR)) was 59.3 (33.5, 93.6) mmol/L. Postoperatively it increased to 115.7 (75.9, 140.6) mmol/L on admission to the PICU, peaked at 131.5 (98.8, 158) mmol/L at 6 h and then decreased thereafter (Figure 3).
The standard base excess stayed within the normal range for the remainder of the time points (Supplementary Table 1). Serum albumin increased from preoperative levels to admission to the PICU and decreased subsequently over the next time points.
The arterial pH decreased slightly towards 24 h but did not drop significantly below 7.35. The arterial partial pressure of carbon dioxide (pCO2) remained within normal limits and the urinary pH remained between 5.5 and 6.5 for the remainder of the time points.
Twenty-one patients (30%) developed AKI. Seven (10%) met AKI stage I criteria, three (4%) met AKI stage II criteria, and 11 (16%) met the definition for AKI stage III. The urinary chloride excretion was associated with kidney injury only at the 24-h time point (Supplementary Table 2). No other parameter was associated with kidney injury at any of the other time points.
The urinary and plasma glutamine levels decreased from admission onwards, with the biggest reduction demonstrated between the pre-surgery and post-surgery time points (Supplementary Table 2).
Discussion
To date, no treatment for AKI, other than supportive measures (such as maintaining adequate perfusion pressure and fluid status) and avoiding nephrotoxins (such as antimicrobials, contrast agents, etc.), has been established. Chloride has become an electrolyte of kidney research, as high administered amounts may lead to AKI. At the same time, high plasma chloride concentrations may be the result of AKI, possibly due to poor excretion. Chloride, or the renal metabolism of chloride, may therefore offer treatment approaches to AKI. We observed in a small study that elevated plasma chloride concentrations may be due to a chloride excretion deficit, rather than because of increased administration. 20 Our study supports these earlier findings that during critical illness (post CPB) renal chloride excretion may be impaired, that this impairment can be greater in those children who develop AKI, and that intravenous chloride administration—while possibly contributing to hyperchloraemia—may not be the primary reason for plasma hyperchloraemia during AKI.
The decrease in renal chloride excretion we observed is associated with a distinct temporary increase in the urinary SID, which decreases as the chloride excretion reduction abates. This urinary SID increase may suggest impaired urinary ammonium excretion as the cause for the reduction in chloride excretion. When measured directly, however, the urinary ammonium did not demonstrate a pattern that correlated with either the urinary chloride or the urinary SID measurement.
The plasma chloride showed a steady rise over the measured time points while the urinary chloride fell below baseline during the first 24 h, which suggests that chloride excretion, rather than an excess in chloride intake, was the cause for the plasma chloride increase. The reduction in urinary chloride excretion is even more striking given that the serum albumin increased during the first 24 h, which normally leads to a metabolic acidosis and a compensatory increase in renal chloride excretion. 13 Therefore, increased urinary chloride excretion is expected with both increased plasma chloride and serum albumin, but neither was demonstrated in our study, suggesting significantly impaired renal chloride excretion. Diuretics did influence the plasma chloride concentration with a decrease almost back to preoperative plasma chloride levels by day 3 in patients receiving them. We deliberately included patients on diuretics in the study because we hypothesised that the renal response to critical illness may be independent of diuretics use—which we demonstrated in the first 24–48 h postoperatively.
The plasma albumin was increased after return from theatre, likely due to albumin that was given as part of the CPB prime. Arguing with Stewart’s approach to blood gas analysis, an increase in albumin will effect a metabolic acidosis which may be compensated for by an increase in chloride excretion, which widens the SID. 29 The albumin peaked on admission to the PICU and fell progressively until the 72-h time point. The chloride excretion on the other hand was minimal at 6 and 12 h and rose to the same level as on admission to the PICU at 24 h. To compensate for the albumin changes chloride excretion should have been elevated in the initial postoperative period, and should have decreased as the albumin fell. The opposite was observed. Had the chloride changes reflected the albumin changes then the chloride excretion should have further fallen in the time periods towards 72 h to compensate for the metabolic alkalosis seen with a fall in albumin but, as in the initial postoperative period, the opposite effect was observed. The chloride balance was maximally positive at 6 h post PICU admission and remained positive over the next days, with an increase in plasma chloride, which should have triggered an increase in urinary chloride excretion. Instead, a decrease in chloride excretion was observed. Finally, hypercapnia can lead to an increase in chloride excretion. 30 However, the pCO2 in our cohort was largely unchanged over the time points and hence does not serve as an explanation for the observed alterations in chloride excretion.
Renal excretion of (excess) chloride normally occurs through an increase of urinary ammonium (binding with chloride) which is produced by the ICCD.12,31,32 Kidney ammoniagenesis begins in the proximal tubule: ammonia binds protons to become ammonium which in turn binds chloride ions, to be excreted in the urine as ammonium chloride.12,31,32 The ammonium concentration in the urine can either be measured directly as an ion, or indirectly as the urine anion gap, defined as [Na] + [K]−[Cl], where
Chloride is freely filtered at the glomerulus, with high tubular reabsorption. The decrease in chloride excretion could therefore result from either a decrease in glomerular filtration (either due to a decrease in renal perfusion due to a low cardiac output state post CPB or tubuloglomerular feedback), or a tubular defect. 37 While the findings of an increase in urinary SID could suggest a tubular defect such as a ‘temporary renal tubular acidosis’, the rapid changes in chloride excretion over the time points would also fit with a variation in glomerular filtration of chloride. The absolute and fractional excretion of sodium changed very little over the time points, which may point towards a glomerular, rather than a tubular, process. We attempted to correlate illness severity post CPB (as a possible index of poor renal perfusion leading to a decrease in glomerular filtration) with chloride excretion, but the number of patients was too small to arrive at definitive results. To resolve the discrepancy between the urinary SID, which would be consistent with a tubular defect, and the fractional excretion rates, which would be consistent with a variation in glomerular filtration rate, seems impossible within the data we have collected.
Our results show a weak association between plasma chloride and AKI. This is consistent with many reports demonstrating the same findings, although none describe how renal chloride excretion is related to AKI.1,2,5,8 –10,38 –40 It remains unclear whether hyperchloraemia is the cause for or the effect of AKI. Acute kidney injury may well lead to hyperchloraemia, which in turn may exacerbate AKI further: both mesangial cell and the juxtaglomerular apparatus react to the elevated plasma chloride concentration by increased excretion of endothelin and angiotensin II, which in turn leads to afferent arteriole constriction and decreased kidney blood flow.41 –43 Urinary chloride excretion was not associated with markers of AKI, possibly due to the fact that criteria for AKI may not detect kidney injury well after CPB surgery.44,45 Creatinine rises slowly during CPB-induced kidney injury, and urine output is often reduced after CPB due to factors other than kidney injury. In addition, large volume fluid administration after CPB surgery as well as third spacing may reduce plasma creatinine levels.45 –47 Finally, urinary electrolytes may be altered due to a large amount of electrolyte administration such as magnesium or potassium chloride, as clinicians may aim for higher levels in cardiac intensive care patients to prevent arrythmias. In combination these circumstances could have contributed to the fact that only plasma chloride at 24 h and none of the other parameters we measured were associated with AKI.
There are several limitations to our study. Most notably, we included patients who received diuretics. We deliberately took this step to assess whether the chloride patterns may be independent of diuretic administration—which they were to an extent. Another limitation is the decline in numbers in the study over time. The median intensive care stay in our unit is 1.9 days, with a significant proportion of patients staying less than 24 h. Therefore, time points beyond 24 h had decreased datapoints, possibly decreasing the accuracy of our observations. We only included very young patients, and those after CPB. To establish how much our results may be applied to the general paediatric intensive care population, or even adult patients, our study would need to be repeated for a larger cohort of general (paediatric or adult) patients. Congenital renal disease may have influenced the results; however, there were no patients with structural renal abnormalities or high preoperative creatinine values identified in our study. While urinary pH changes can lead to urinary SID changes this is an unlikely explanation for our observations, given that the urinary pH varied very little. Finally, antidiuretic hormone (ADH) can increase renal chloride excretion. 48 However, post CPB surgery ADH is, if anything, increased, rather than decreased, which should lead to an increase, rather than the (in our study) observed decrease in urinary chloride excretion. 49
We showed that both urinary as well as plasma glutamine concentrations decreased over the course of the study, with the urinary glutamine stabilising at 12 h. It would have been informative to see urinary glutamine measurements beyond 24 h. For practical and cost reasons this was not possible. There are several recent animal studies on the effect of glutamine administration on renal injury,50 –52 demonstrating a positive effect. Our findings would fit with these physiology-based studies, although treatment of critically ill patients with glutamine would need to be targeted to those with renal injury, given that glutamine may have a negative effect when given prophylactically to any critically ill patient.53,54
We have not compared the electrolyte and glutamine concentrations with other biomarkers that are predictive of AKI. However, it remains unclear which of these biomarkers (neutrophil gelatinase-associated lipocalin, macrophage inhibitory cytokine 1 and others) are truly predictive of kidney injury.55 –58 Moreover, due to ethical constraints we were unable to collect more patient blood for additional analyses.
In summary, our study suggests that hyperchloraemia in a critical illness state may be related to a chloride excretion deficit which may reflect initial stages of kidney impairment. The decrease in urinary chloride excretion correlated with AKI. Targeting renal chloride metabolism, possibly with the administration of glutamine as has been described before but not instituted more widely, rather than just limiting intravenous chloride administration, may therefore offer a treatment approach to AKI.
Supplemental Material
sj-pdf-1-aic-10.1177_0310057X241265119 - Supplemental material for Urinary chloride excretion in critical illness and acute kidney injury: a paediatric hypothesis-generating cohort study post cardiopulmonary bypass surgery
Supplemental material, sj-pdf-1-aic-10.1177_0310057X241265119 for Urinary chloride excretion in critical illness and acute kidney injury: a paediatric hypothesis-generating cohort study post cardiopulmonary bypass surgery by Adrian C Mattke, Kerry E Johnson, Krishanti Ariyawansa, Peter Trnka, Prem S Venugopal, David Coman, Andreas Schibler and Kristen Gibbons in Anaesthesia and Intensive Care
Supplemental Material
sj-pdf-2-aic-10.1177_0310057X241265119 - Supplemental material for Urinary chloride excretion in critical illness and acute kidney injury: a paediatric hypothesis-generating cohort study post cardiopulmonary bypass surgery
Supplemental material, sj-pdf-2-aic-10.1177_0310057X241265119 for Urinary chloride excretion in critical illness and acute kidney injury: a paediatric hypothesis-generating cohort study post cardiopulmonary bypass surgery by Adrian C Mattke, Kerry E Johnson, Krishanti Ariyawansa, Peter Trnka, Prem S Venugopal, David Coman, Andreas Schibler and Kristen Gibbons in Anaesthesia and Intensive Care
Supplemental Material
sj-pdf-3-aic-10.1177_0310057X241265119 - Supplemental material for Urinary chloride excretion in critical illness and acute kidney injury: a paediatric hypothesis-generating cohort study post cardiopulmonary bypass surgery
Supplemental material, sj-pdf-3-aic-10.1177_0310057X241265119 for Urinary chloride excretion in critical illness and acute kidney injury: a paediatric hypothesis-generating cohort study post cardiopulmonary bypass surgery by Adrian C Mattke, Kerry E Johnson, Krishanti Ariyawansa, Peter Trnka, Prem S Venugopal, David Coman, Andreas Schibler and Kristen Gibbons in Anaesthesia and Intensive Care
Footnotes
Author contribution(s)
Data are available on reasonable request
Deidentified participant data are available by email request from Adrian Mattke. A data sharing agreement will need to be put in place for the data to be shared. The Human Research Ethics Committee at Queensland Children’s Hospital will need to approve data sharing agreements. Reuse may be permitted should the Human Research Ethics Committee approve this.
Declaration of conflicting interests
The author(s) have no financial or non-financial interests or benefits related to the work published in this article to declare.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article. The study was supported in kind by the Queensland Children’s Hospital Paediatric Intensive Care Unit.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.