
Abstract
Some degree of exertional rhabdomyolysis (ER), striated muscle breakdown associated with strenuous exercise, is a well-known phenomenon associated with endurance sports. However in rare cases, severe and/or recurrent ER is a manifestation of an underlying condition, which puts patients at risk for significant morbidity and mortality. Selecting the patients that need a diagnostic work up of an acute rhabdomyolysis episode is an important task.
Based on the diagnostic work up of three illustrative patients treated in our hospital, retrospectively using the ‘RHABDO’ screening tool, we discuss the clinical and biochemical clues that should trigger further investigation for an underlying condition. Finally, we describe the most common genetic causes of this clinical syndrome.
Introduction
Exertional rhabdomyolysis (ER) is a frequent syndrome treated by a wide variety of medical disciplines. For these different specialties, it is often difficult to distinguish a normal physiological response to exercise—including muscle soreness and an elevated blood creatine kinase (CK) level—from a pathological and clinically more relevant response in relation to the type of exercise. The latter again includes muscle soreness (albeit more often of a more severe and/or delayed type), fever, and biochemical evidence of rhabdomyolysis.
The distinction between both responses is nevertheless important as a patient having an underlying genetic condition is prone to recurrence and should therefore be warned about the risk. Consequently, clinical and biochemical findings have to be interpreted adequately, and clues for further non-invasive and invasive investigations defined.
A recent review article aimed to define the triggers for further investigation, and a screening tool under the acronym ‘RHABDO’ was proposed. 1 Even though this practical tool is available, all too often ER necessitating hospital admission is not properly investigated for an underlying cause, even when several clues warrant this to be the case.
In this article, we review the diagnostic work up in three illustrative cases treated at our institution by applying this acronym, and present a summary of the most common underlying causes of this clinical syndrome.
Written informed consent was obtained from all participating patients and the study was approved by the institutional review board.
Case descriptions
The following three patients were treated for severe ER by a multidisciplinary team of emergency physicians, intensive care specialists, neurologists, and the malignant hyperthermia (MH) laboratory of the University Hospital of Antwerp in Belgium.
Case 1
Male, 21 years of age. In December 2016, at the age of 18 years, the patient presented with the complaint of severe cramping pain after having played soccer, even though he had not experienced the game as intense. Outside temperature was around 5°C. At admission to the emergency department, his CK level exceeded 45,000 IU/L and myoglobinuria was found. He stayed in the hospital for one week and no complications occurred. A second episode was noted in March 2018 during a tournament, again in normal ambient temperatures, in which he played several matches on consecutive days. Myalgia was pronounced but less severe than in 2016; his CK level was 7500 IU/L. Triggers other than physical effort could not be identified.
Neurological examination was normal. Blood tests showed a basal CK level of 99 IU/L (N 30–200 IU/L). Muscle magnetic resonance imaging (MRI) was normal.
The patient had been exposed to volatile anaesthetics on several occasions without any adverse reaction. The family history was negative for signs of myopathy and anaesthesia-related events.
Muscle biopsy performed three months after his last episode of ER showed normal histological findings. In vitro contracture testing for MH susceptibility according to the protocol of the European Malignant Hyperthermia Group (EMHG) 2 was within normal limits, ruling out MH susceptibility. Molecular genetic analysis could not reveal any mutation in RYR1, nor in the carnitine palmitoyl transferase 2 gene (CPT2).
Case 2
Male, 56 years of age. This patient presented with myalgia after exercise described as a sensation of severe tension or cramping pain, unilateral or bilateral. The complaints were not related to the type of effort as the pain could be elicited by only short exercise and was not necessarily provoked by longer exercise. CK value increased to 14,000 IU/L during one myalgia episode. For years, he had been an amateur cyclist accustomed to regular hour-long exercise. His complaints started at the age of 40. There was no previous exposure to volatile anaesthetics. Family history was negative for similar types of complaints or anaesthesia-related events.
Clinical examination was normal. Basal CK value was variably increased (269 to 1771 IU/L); lactate was 1.2 mmol/L. Ischaemic forearm testing was normal and muscle MRI could not detect any alterations in muscle signal intensity. A muscle biopsy showed increased variability in fibre size and an increased number of internal nuclei. The in vitro contracture test (IVCT) was indicative of MH susceptibility (5 mN contracture at 2 mM caffeine and 14 mN contracture at 2 vol% halothane). CTP2 sequencing was negative, but a heterozygous missense mutation in exon 43 of RYR1 was identified (p.Asn2342Ser) (this clinical case has previously been reported in a paper focusing on the possible risk of administration of volatile anaesthetics in patients with ER3).
Case 3
Female, 22 years of age. She was first investigated at the age of 20 years, presenting with myalgia of the lower limb muscles for two days and clinical myoglobinuria. Neurological examination was unremarkable, particularly regarding manual strength testing, apart from tenderness of lower limb muscles. CK level was 82,324 IU/L. She was admitted to the hospital and intravenous hyperhydration was started. Recovery was uneventful.
Retrospectively, she had experienced multiple episodes of muscle soreness and stiffness since the age of ten years, when she manifested these symptoms for the first time during a mountain walk. At the age of 14 years, she had had similar complaints during ballet training. However, since then, these episodes had also occurred in conjunction with viral upper respiratory tract infections, stress (e.g. exams), skipping meals and, as before, accustomed yet relatively prolonged exercise of about one hour, even at normal or low ambient temperatures.
Basal CK level was within normal limits. Plasma acylcarnitine profiling in the acute phase of the rhabdomyolysis showed low acetylcarnitine (C2). Muscle MRI did not reveal any structural abnormalities. Family history was negative with regard to muscle symptoms and anaesthesia-related problems. Sanger sequencing of CPT2 revealed the most recurrent mutation, p.Ser113Leu, in homozygosity. In view of this diagnosis, muscle biopsy and in vitro contracture testing for MH susceptibility were not indicated.
Discussion
When dealing with ER, as in the cases presented, three main questions have to be considered: first, is the observed degree of rhabdomyolysis in relation to the effort and circumstances or not; second, is there a risk of recurrence; and third, is there a risk of other complications, most notably anaesthesia-related MH?
A ‘physiological’ or ‘expected’ response of muscle to exercise means either a minor to moderate degree of muscle damage following strenuous exercise or a more pronounced degree of rhabdomyolysis following extreme or unaccustomed exercise. On the other hand, muscle breakdown that occurs during or following accustomed exercise in normal environmental conditions with well-trained individuals has to be regarded as an ‘unexpected’ or ‘pathological’ response. However, as terms such as ‘minor’, ‘moderate’, ‘strenuous’, ‘unaccustomed’ and ‘delayed’ are prone to subjective interpretation, the distinction is hard to make.
Over the last few years, consensus has grown as to what should be the definition of ER, a concept that encompasses three key features:
A CK increase peaking at three to four days, followed by normalization within several weeks. The CK increase is preceded by strenuous exercise; alternatively by accustomed exercise in abnormal environmental circumstances. The CK increase incurs a clinical correlate of myalgia, swelling and/or weakness of muscle (either generalized or in particular groups of muscle depending on the type of exercise).
1
It is well known that blood CK levels increase considerably in the days following prolonged exercise and remain elevated for several days. Of the 203 participants subjected to 50 maximal eccentric contractions of the elbow flexor muscles, 111 had CK values at four days post-exercise of over 2,000 IU/L, and 51 exceeded 10,000 IU/L. At day 10 after the exercise, CK levels were still 300% above baseline. 4 As the upper limits of normal levels already vary considerably between laboratories depending on the technique of measurement and ethnicity, 5 the matter is further complicated.
CK values felt to be indicative of an abnormal degree of rhabdomyolysis have been variably reported as: 5–50 times the upper limit, >1000 IU/L up to >10,000 IU/L. However, one cannot rely solely on CK levels to differentiate a physiological exertional CK elevation from rhabdomyolysis based on an underlying cause. Indeed, other elements have to be taken into consideration and, with this aim in mind, a screening tool with the acronym ‘RHABDO’ was published in 2016 by Scalco et al.
1
The following clues may point to an underlying genetic disorder:
R: Recurrent episodes of ER H: HyperCKaemia persists eight weeks after the event A: Accustomed physical exercise B: Blood CK > 50× upper limit of normal (>10,000 IU/L in female Caucasian patients) D: Drugs/medication/supplements and other exogenous and endogenous factors cannot sufficiently explain the rhabdomyolysis severity O: Other family members affected/other exertional symptoms (cramps or myalgia).
Applying this acronym tool to our patients resulted in at least four risk factors warranting further investigation being positive, whereas in fact a single one would suffice. This is illustrated in Table 1.
‘RHABDO’ screening application.
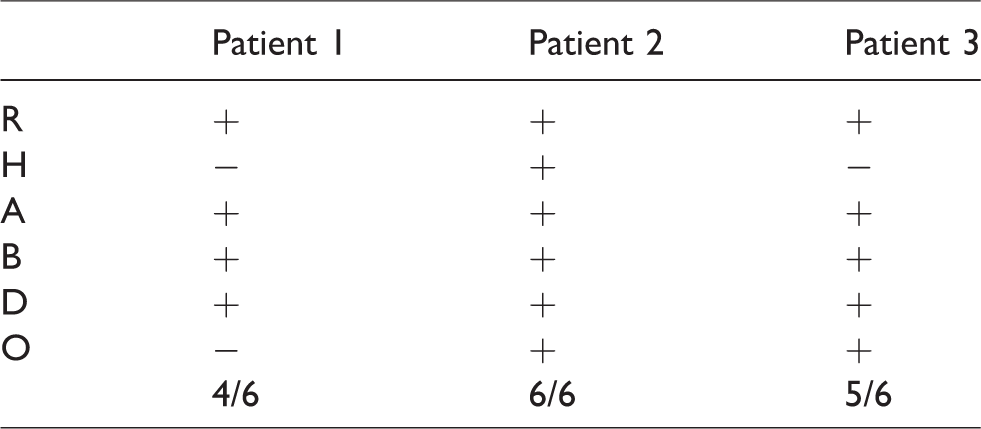
The risk of ER recurrence is real if an underlying genetic condition can be identified. Although many of these are very rare, some inherited causes of rhabdomyolysis are more prevalent.
Even though the sickle cell trait has a very limited effect on the physiological response to exercise, a greater incidence of ER has been reported in such patients over several decades. 6 This has recently been confirmed in a population-based, longitudinal study conducted in US Army soldiers. A significantly higher risk of ER was identified among black recruits with the sickle cell trait than among those without the trait. 7 In the US, 7%–10% of the African population has this trait. If the sickle cell trait is an underlying cause, then muscle damage is triggered by effort and has a variable time of onset: early, late or after the event. The major diagnostic clue in such cases is being of African descent.
Secondly, a number of inherited myopathies predispose to ER. However, these are highly heterogeneous and it is felt likely that polygenetic factors lead to the huge variance in the degree of rhabdomyolysis in particular circumstances of vigorous exercise. 8 Algorithms for both a clinical diagnostic approach 9 and the search for a genetic defect 10 have been developed, and are very useful.
The inherited neuromuscular disorders associated with episodes of adult-onset ER are classified into the following categories: disorders of intramuscular calcium release and excitation–contraction coupling, disorders of glycogen metabolism, disorders of fatty acid metabolism, muscular dystrophies, mitochondrial disorders and miscellaneous. 10
The ‘most prevalent’ or ‘least rare’ myopathies are RYR1-related disorders. RYR1 encodes the principal sarcoplasmic reticulum calcium release channel and plays a crucial role in excitation–contraction coupling. RYR1 missense mutations have been found in 50%–60% of families with proven MH. 11 The possible link between ER and MH susceptibility has been explored in a number of studies over the last few years. A first large series of 39 unrelated families with rhabdomyolysis and/or exertional myalgia was published in 2013. 12 Nine heterozygous mutations were reported in 14 families, five of which had previously been associated with MH. Since then, it has been shown in different cohorts of patients with ER that causative mutations or potentially pathogenic variants in RYR1 and CACNA1S account for a substantial proportion of patients presenting with unexplained rhabdomyolysis and/or exertional myalgia.13–15
It appears that this genetic predisposition in combination with various stressors (e.g. inhalational anaesthesia, viral infections, environmental heat, strenuous exercise, severe stress and/or drugs) can elicit acute rhabdomyolysis. Partly based on these findings, the US Department of Defense defines MH as a disqualifying condition to their fitness for duty determinations. 16
The second case illustrates the complex relationship between ER, histopathology, MH susceptibility and RYR1 analysis. This patient’s MH susceptibility status was assessed and confirmed by in vitro contracture testing with halothane and caffeine. The test is considered positive if a sustained contracture of at least 2 mN is obtained at caffeine concentrations ≤ 2 mM, and/or halothane concentrations of 2 vol% or less. Normal individuals do not react at the threshold concentrations of either agent. The test sensitivity is 99% and its specificity is 93.5%. 17 Even though the p.Asn2342Ser variant (previously reported in Italian families 18 ) is not classified as a true diagnostic mutation in the EMHG genetic database (https://www.emhg.org/diagnostic-mutations; accessed January 30 2019), there is corroborative experimental evidence that this mutation can be used for predictive genetic testing. Indeed, the 4-chloro-m-cresol-induced proton release rate of lymphoblastoid cells—as an indirect indicator of activation of the RYR1 calcium channel—was shown to be increased in the RYR1Asn2342Ser mutated channels (and inhibited by dantrolene). 18 In this patient, the positive IVCT test suggested additional evidence for pathogenicity. Therefore, we concluded that he was clearly at risk for recurrent ER and MH, in all likelihood on the basis of an RYR1 mutation with ‘gain of function’ of the calcium release channel of the sarcoplasmic reticulum. As MH is considered to be an autosomal dominant disorder, his family members also are at risk for this potentially life-threatening anaesthesia complication. The histopathological changes as such are non-specific but nevertheless consistent with a RYR1 myopathy. Patient 1 is at risk for recurrent ER, but not at risk for MH during anaesthesia. An underlying condition could not be identified. The familial implications remain unknown.
Regarding disorders of glycogen metabolism, the most common disease in this group is glycogen storage disease type V, McArdle’s disease. The trigger for rhabdomyolysis is exercise with an early symptom onset, which is within minutes. Therefore, these are not the type of patients that will present with rhabdomyolysis at the end of endurance exercise as described in these cases. Baseline CK levels are high.
Other categories include:
Disorders of fatty acid metabolism. Carnitine palmitoyl transferase deficiency due to autosomal-recessive mutations in the CPT2 gene is the most common disorder in this group. The severity of the symptoms differs according to the degree of reduction in enzyme activity, with the more severe forms expressed as fatal neonatal or infantile-onset variants, and the less severe expressions as adult-onset forms with exercise intolerance, myalgia and recurrent ER. In the latter group, prolonged (one hour) exercise but also exposure to heat, infection, fasting and stress can provoke a crisis. Baseline CK levels are normal. CPT2 deficiency was found to be the cause in the third patient. A second disorder of fatty acid metabolism is a deficiency of very-long-chain acyl-CoA dehydrogenase. The muscular dystrophies are usually diagnosed because of progressive weakness, but there is an increased susceptibility for ER that may be the presenting problem. Baseline CK level is high. Mitochondrial DNA mutations associated with ER are rare but have been reported in cases with mutations in the cytochrome b and cytochrome c oxidase genes. Early and excessive fatigue is a major physical complaint, which is not the case for the other groups, and which was not a complaint in the patients investigated.
Defining a general diagnostic approach to ER in adults is difficult in view of the multiple potential genetic causes. The following algorithmic approach to clinical decision-making can be used to differentiate the more prevalent underlying myopathies (Figure 1).
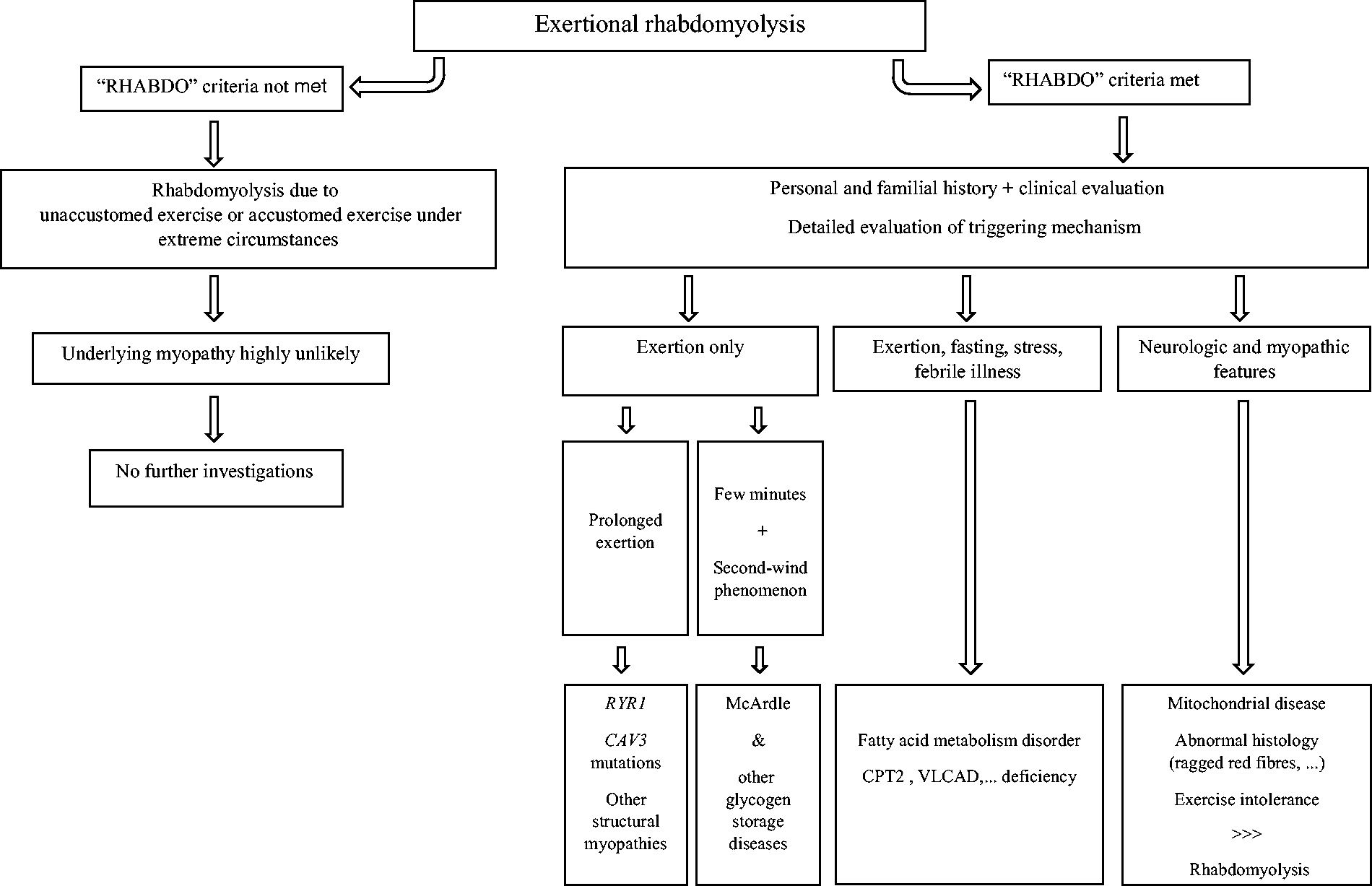
Flowchart presenting a clinical pattern recognition approach to patients with exertional rhabdomyolysis.
Baseline investigations include identification of the triggers for rhabdomyolysis as well as the relative timing of onset, neurological examination, and serial CK and myoglobinuria measurements.
Second-phase tests include non- or minimally invasive tests such as fasting acyl-carnitine profiling, electromyography, ischaemic forearm testing and MRI. Analysis of T1-weighted MRI images of pelvis, thigh and leg muscles demonstrates significant differences in the pattern of affected and spared muscles for a number of myopathies, such as the muscular dystrophies and RYR1 myopathies.19,20 This may assist in the prioritization of genetic testing of particular genes such as RYR1.
If this is not the case, third-phase investigations such as muscle biopsy may help in targeting genetic testing and the most appropriate ‘myopathy/rhabdomyolysis DNA sequencing panel’. The analytical sensitivity of sequencing individual genes in patients with specific clinical features, abnormal metabolites, and/or histo-enzymatic deficiency is high; whereas this is much less the case if no such presumptive diagnosis can be made.
In a significant proportion of patients presenting with ER the precise underlying mechanism remains unsolved.
Footnotes
Acknowledgements
Author contributions: KH is the main author of the manuscript. WD and JD provided information about the different cases and reviewed the manuscript. LH and JB designed the aim of the manuscript and acted as co-writers.
Declaration of conflicting interests
The author(s) have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.