
Abstract
NSG-SGM3 and NOG-EXL mice combine severe immunodeficiency with transgenic expression of human myeloid stimulatory cytokines, resulting in marked expansion of myeloid populations upon humanization with CD34+ hematopoietic stem cells (HSCs). Humanized NSG-SGM3 mice typically develop a lethal macrophage activation syndrome and mast cell hyperplasia that limit their use in long-term studies (e.g., humanization followed by tumor xenotransplantation). It is currently unclear to what extent humanized NOG-EXL mice suffer from the same condition observed in humanized NSG-SGM3 mice. We compared the effects of human CD34+ HSC engraftment in these two strains in an orthotopic patient-derived glioblastoma model. NSG-SGM3 mice humanized in-house were compared to NOG-EXL mice humanized in-house and commercially available humanized NOG-EXL mice. Mice were euthanized at humane or study endpoints, and complete pathological assessments were performed. A semiquantitative multiparametric clinicopathological scoring system was developed to characterize chimeric myeloid cell hyperactivation (MCH) syndrome. NSG-SGM3 mice were euthanized at 16 weeks after humanization because of severe deterioration of clinical conditions. Humanized NOG-EXL mice survived to the study endpoint at 22 weeks after humanization and showed less-severe MCH phenotypes than NSG-SGM3 mice. Major differences included the lack of mast cell expansion and limited tissue/organ involvement in NOG-EXL mice compared to NSG-SGM3 mice. Engraftment of human lymphocytes, assessed by immunohistochemistry, was similar in the two strains. The longer survival and decreased MCH phenotype severity in NOG-EXL mice enabled their use in a tumor xenotransplantation study. The NOG-EXL model is better suited than the NSG-SGM3 model for immuno-oncology studies requiring long-term survival after humanization.
Keywords
Cell/tissue-based humanized mice are interspecies chimeras resulting from the functional integration and development of human-into-mouse xenotransplants. 6 These models are generated using severely immunodeficient mouse recipients that lack key components of both innate and adaptive immunity, allowing the engraftment of a wide range of human cells/tissues. 19 Ideally, the transplanted human cells/tissues should recapitulate, as much as possible, the same biological features observed in the donor.6,29
NSG-SGM3 and NOG-EXL mice combine the severe immunodeficiency caused by loss-of-function mutations of the Prkdc and Il2rg genes on a nonobese diabetic (NOD) background with the transgenic expression of human myeloid stimulatory cytokines, including granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-3 (IL-3), and KIT ligand (also known as steel factor or stem cell factor), the latter featured only in the NSG-SGM3 model.5,28 Because of these characteristics, both models have been reported to be ideal recipients for humanization with human CD34+ hematopoietic stem cells (HSCs), allowing improved multilineage reconstitution and function of hematolymphoid cells with robust expansion of myeloid populations.9,7,12,27
However, there are limitations that restrict the utility of these mice in long-term experiments. Specifically, at around 12–18 weeks after human CD34+ HSC engraftment, NSG-SGM3 mice invariably develop a lethal phenotype characterized by the expansion and hyperactivation of the human myeloid cell compartment, which is driven by the supraphysiologic expression levels of the transgenic human cytokines.10,13,23,28,30 This complex phenotype involves multiple myeloid cell lineages combining features of hemophagocytic macrophage activation syndrome (MAS)—or hemophagocytic lymphohistiocytic (HLH)-like disease10,23,27,28,30—and mast cell/eosinophil hyperplasia. 10 For this reason, we decided to adopt chimeric myeloid cell hyperactivation (MCH) syndrome as a more general term, which combines all the manifestations associated with this entity. The early mortality due to this disorder greatly limits the use of this model in long-term studies, especially those requiring humanization followed by tumor xenotransplantation. 23
While there is evidence of much longer-term survival and model stability in humanized NOG-EXL mice,13,25 it is currently unclear to what extent these animals suffer from the same conditions observed in the NSG-SGM3 mouse line. Therefore, the goal of this longitudinal study was to compare the effects of human CD34+ HSC engraftment in NSG-SGM3 and NOG-EXL mice for the development of an orthotopic patient-derived xenograft model of intracranial glioblastoma. This investigation focused on the comparative assessment of the onset, progression, and severity of disorders resulting from MCH syndrome, including MAS and mast cell/eosinophil hyperplasia.
Materials and Methods
Animals and Experimental Design
All the mice included in this study were maintained by the Stem Cell and Xenograft Core (RRID: SCR_010035) within the Perelman School of Medicine at the University of Pennsylvania under the protocol 803506, which has been reviewed and approved by the Institutional Animal Care and Use Committee. The NSG-SGM3 (NOD. Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3, CSF2, KITLG)1Eav/MloySzJ) colonies were established from animals originally purchased from Jackson Laboratories (Bar Harbor, ME). NOG-EXL (NOD. Cg-Prkdcscid Il2rgtm1Sug Tg(SV40/HTLV-IL3,CSF2)10-7Jic/JicTac) mice were obtained from Taconic Biosciences (Rensselaer, NY) at 4 weeks of age and maintained in the Stem Cell and Xenograft Core. At 8 weeks of age, 10 female NSG-SGM3 and NOG-EXL mice were preconditioned with 30 mg/kg intraperitoneal busulfan and, within 24 hours, received intraosseous administration of 2 × 104 bone marrow–derived human CD34+ HSCs. Mononuclear cells were isolated by Ficoll density centrifugation (Ficoll-Paque Premium Fisher 45-001-751, Hampton, NH) followed by ammonium chloride (Stem Cell Technologies 07850, Vancouver, Canada) treatment for red blood cell lysis. Mononuclear cells were submitted to CD34+ selection using Miltenyi Biotec magnetic microbeads (130-046-702) and an Automacs Pro. The initial aim of the study was to establish a patient-derived orthotopic glioblastoma model in both NSG-SGM3 and NOG-EXL mice humanized with autologous CD34+ HSCs isolated from the bone marrow of the same tumor donor. However, the amount of autologous CD34+ HSCs was not sufficient to allow the humanization of both mouse groups. Based on our previous experience, regardless of the autologous or allogeneic source of HSC, humanized NSG-SGM3 mice invariably develop lethal MCH syndrome before reaching the tumor transplantation timepoint. 23 Therefore, autologous engraftment was prioritized in NOG-EXL mice, where early lethality upon humanization has never been documented. Female mice were used in this study due to the known stronger engraftment of human CD34+ HSCs in female NSG mice, to maximize the chance of engraftment of the limited supply of single-donor-derived cells.14,16,26 An allogeneic source of CD34+ HSCs isolated from a single fetal liver donor was instead used for the humanization of NSG-SGM3 mice. To ensure that the mice reached a minimum 5% human leukocyte chimerism by flow cytometry (see section below on flow cytometry), peripheral blood was collected from the retro-orbital sinus under 1%–4% isoflurane general anesthesia at 10 weeks after humanization.
Twelve commercially available humanized NOG-EXL (huNOG-EXL, HSCCB-13395-F) female mice were obtained from Taconic Biosciences at 15 weeks after humanization (approximately 20 weeks of age). The protocol used by Taconic Biosciences to humanize these mice is considered proprietary. Therefore, specific details regarding conditioning as well as source, amount, and route of administration of CD34+ HSCs were not disclosed. Preconditioned female NSG-SGM3 and NOG-EXL mice (5 of each strain) without administration of human CD34+ HSCs were also included in the study as control animals.
Glioblastoma tissue was obtained from the same adult donor patient whose CD34+ HSCs were used to humanize one of the NOG-EXL groups, as described earlier. Organoids were generated from the tumor biopsy as previously described. 8 A total of 3.25 × 104 glioblastoma organoid cells were injected intracranially into the left hemisphere (2 mm lateral and 1 mm anterior to the lambda) at 17 weeks after humanization. The consent to use cancer patient/human donor-derived specimens for research was obtained under a protocol approved by the University of Pennsylvania’s Institutional Review Board (IRB protocol 836366).
Clinical conditions and body weight measurement of the mice were assessed biweekly throughout the experiment. In addition, clinical monitoring was performed daily (for at least 3 consecutive days) after blood collection and intracranial tumor transplantation. NSG-SGM3 mice were monitored every other day starting from week 12 after humanization. Loss of more than 20% of body weight and/or development of severe clinical signs such as lethargy, hunched posture, and labored breathing were the criteria used for humane euthanasia.
A schematic overview of the experimental design and main study outcome(s) is provided in Supplemental Figure S1.
Pathological Assessment
When mice reached 22 weeks after humanization (5 weeks after tumor xenotransplantation) or humane endpoints, they were euthanized by carbon dioxide asphyxiation. A complete necropsy with macroscopic examination was performed. The following organs/tissues were collected and fixed in 10% neutral-buffered formalin for histopathologic assessment: whole head (brain, eyes and adnexa, oral cavity, nasal passages, ears, and pituitary gland), skin (dorsal region, auricle, and muzzle), salivary glands with cervical lymph nodes, larynx, trachea, esophagus, lungs, heart, liver, pancreas, kidneys, adrenal glands, gastrointestinal tract (stomach, duodenum, jejunum, ileum, cecum, proximal colon, and distal colon/rectum), gonads and reproductive tract, urinary tract, external genitalia, spleen, spine, sternum, mesentery with mesenteric lymph nodes, and quadriceps femoris. The head and sternum were decalcified in a 15% formic acid solution for 24 hours. For 2 humanized NOG-EXL mice from each group, the head was collected for a separate procedure and was not available for histopathologic assessment. Whole body weights and the weights of the spleen, liver, right kidney, left kidney, and heart were recorded during necropsy. Formalin-fixed tissue samples were trimmed according to a standardized approach for rodent studies adapted from the Registry of Industrial Toxicology Animal-data (RITA) guidelines.11,15,20 Trimmed tissue samples were then routinely processed for paraffin embedding, sectioning, and hematoxylin and eosin staining. Prussian blue staining was performed on liver sections, as previously described. 23 The Aperio Versa 200 slide scanner was used for whole-slide image acquisition.
To characterize the clinicopathological impact of transplantation of human CD34+ HSCs on mouse recipients, a scoring system was developed based on histopathologic and hematologic findings consistent with MCH phenotype. Each finding was assigned a semiquantitative score corresponding to minimal (1), mild (2), moderate (3), or severe (4) effects on the tissue, and a cumulative score was calculated for each mouse. Details regarding the scoring criteria for each parameter are listed in Supplemental Table S1. The anemia scoring criteria were based on hemoglobin level and adapted from Raabe et al. 18
Hematology
Terminal blood collection was performed at necropsy by cardiac puncture and preserved in specific ethylenediaminetetraacetic acid (EDTA) anticoagulant-coated tubes (Greiner Bio-One #450480, Monroe, NC). Hematological analysis was performed using an IDEXX ProCyte Hematology Analyzer (IDEXX Laboratories, Columbia, MO).
Immunohistochemistry
For immunohistochemistry (IHC), 5-µm-thick paraffin sections were mounted on ProbeOn slides (Thermo Fisher Scientific, Waltham, MA). The IHC procedure was performed using a Leica BOND RXm automated platform combined with the Bond Polymer Refine Detection kit (Leica #DS9800). Briefly, after dewaxing and rehydration, sections were pretreated with the epitope retrieval BOND ER2 high-pH buffer (Leica #AR9640) for 20 minutes at 98°C. Endogenous peroxidase was inactivated with 3% hydrogen peroxide for 10 minutes at room temperature. Nonspecific tissue-antibody interactions were blocked with Leica PowerVision IHC/ISH Super Blocking solution (PV6122; Leica Biosystems, Deer Park, IL) for 30 minutes at room temperature. The same blocking solution also served as diluent for the primary antibodies. Details regarding primary antibodies are listed in Supplemental Table S2. Primary antibodies were incubated on the sections for 45 minutes at room temperature. A biotin-free polymeric IHC detection system consisting of horseradish peroxidase (HRP)-conjugated anti-rabbit IgG was then applied for 25 minutes at room temperature. Immunoreactivity was revealed with the diaminobenzidine (DAB) chromogen reaction. Slides were counterstained in hematoxylin, dehydrated in an ethanol series, cleared in xylene, and permanently mounted with a resinous mounting medium (Thermo Scientific ClearVue coverslipper). The Aperio Versa 200 instrument was used for image acquisition. Details regarding primary antibodies and IHC procedures are listed in Supplemental Table S2.
Flow Cytometry
Flow cytometry analysis was performed on an LSR II flow cytometer (Becton Dickinson, San Jose, CA, USA). Peripheral blood was stained with a live/dead marker (Fixable Viable Stain 510; BD Biosciences), followed by blocking with CD16/CD32 Fc (BD Biosciences) and labeling with antibodies for cell surface markers: human CD3-BV421 (BD, Cat No: 562426), human CD19-APC (BD, Cat No: 555415), human CD45-PE (BD, Cat No: 555483), mouse CD45-PE-Cy7 (BD, Cat No: 552848), and human CD33-BB-515 (BD, Cat No: 564588). Engraftment levels in the xenotransplanted mice were expressed as the percentage of the human CD45-labeled cells in relation to the total number of live nucleated cells following the formula: [hCD45%/(hCD45% + mCD45%)] × 100.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 9.4.1. (GraphPad Software, Boston, MA). Comparisons of multiple groups were calculated by Kruskal-Wallis test with Dunn’s multiple comparisons test for noncontinuous values and one-way analysis of variance (ANOVA) with Tukey’s test for continuous values. Comparisons of two groups were calculated by unpaired Welch’s t test for continuous data or Mann-Whitney test for noncontinuous data. P < 0.05 was considered significant. Survival was calculated by log-rank (Mantel-Cox) test.
Results
Clinical Course and Survival
None of the 10 humanized NSG-SGM3 mice reached the tumor xenotransplantation timepoint at 17 weeks after human CD34+ HSC delivery (Fig. 1a). One animal was found dead at 15 weeks after humanization, and the remaining animals were sacrificed at 16 weeks due to the development of severe clinical signs such as lethargy, hunched posture, and labored breathing or loss of more than 20% body weight. On the contrary, all but 1 of the 22 humanized NOG-EXL mice appeared clinically normal and reached the terminal endpoint of the experiment, 22 weeks after human CD34+ HSC transplantation including tumor xenotransplantation at 17 weeks (Fig. 1a). One commercially available huNOG-EXL mouse was sacrificed at 20 weeks after humanization due to the manifestation of severe clinical signs and significant weight loss.
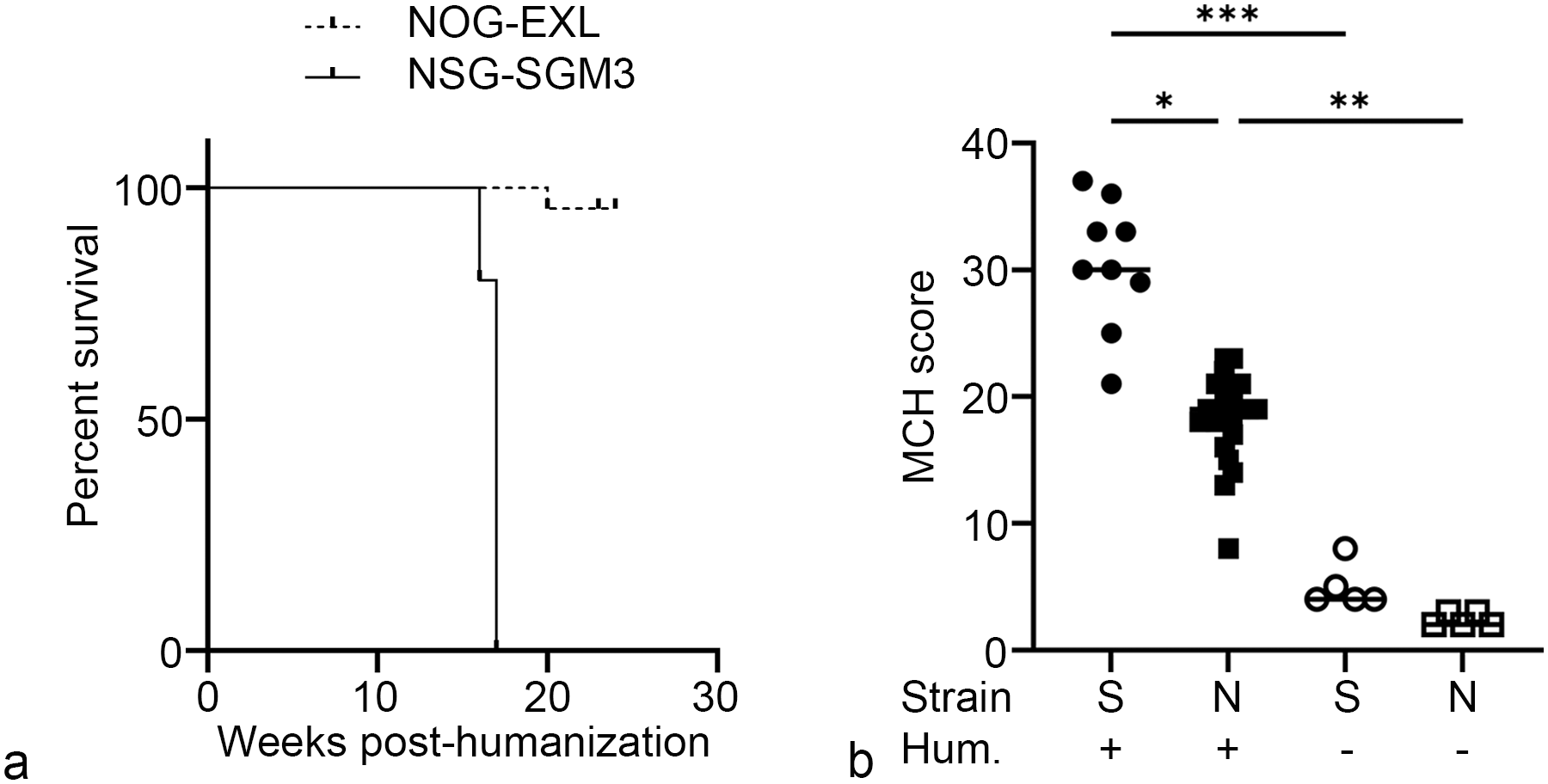
Humanized NOG-EXL mice (combining both the commercially available and the in-house groups) survive to experimental endpoint and exhibit less severe myeloid cell hyperactivation (MCH) syndrome. (a) Kaplan-Meier curve for humanized NOG-EXL and NSG-SGM3 mice. P < .0001 (log-rank [Mantel-Cox] test). (b) Semiquantitative score of the MCH syndrome based on histopathologic and hematologic findings collected at necropsy. S, NSG-SGM3; N, NOG-EXL; Hum., humanized. Kruskal-Wallis test with Dunn’s test. *P < .05, **P < .01, ***P < .001. n = 22 mice/group (humanized NOG-EXL), 9 mice/group (humanized NSG-SGM3), and 5 mice/group (non-humanized).
Macroscopic Pathology
Complete necropsy was performed on all mice considered in the study including animals that were found dead, the NSG-SGM3 mice euthanized for humane reasons at 16 weeks after humanization, and the NOG-EXL mice that reached the terminal endpoint at 22 weeks after humanization. Macroscopic findings in humanized NSG-SGM3 mice included splenomegaly (10/10), an irregular to granular surface of the liver (6/10), unkempt haircoat (6/10), and macroscopically detectable lymph nodes or thymus (3/10). Findings in both groups of humanized NOG-EXL mice were comparable to each other and similar to the humanized NSG-SGM3 mice and included splenomegaly (19/22), an irregular to granular surface of the liver (17/22), unkempt haircoat (1/22), and generalized tissue pallor (1/22).
Histopathology, Semi-quantitative Scoring, and IHC
Histopathologically, all groups of humanized mice exhibited lesions consistent with MCH syndrome, in multiple organs and tissues. However, there were significant quantitative and qualitative differences between the mouse strains, especially regarding lesion severity, composition, and distribution. To characterize the severity of MCH syndrome in these mice, we developed a scoring system based on histopathologic and hematologic findings (Supplemental Table S1). Each finding was assigned a semiquantitative score, and a cumulative score was calculated for each mouse (Fig. 1b and Supplemental Table S3). The humanized NOG-EXL mice were significantly less severely affected (median 19, interquartile range 17–21) than the humanized NSG-SGM3 mice (median 30, interquartile range 27–34.5) despite their much longer post-humanization interval (i.e., 22 weeks in NOG-EXL versus 16 weeks in NSG-SGM3). This difference in MCH scores was primarily ascribable to the absence or decreased severity of lesions in the pancreas, bone marrow, meninges, spleen, and skin, as well as lower leukocyte counts of humanized NOG-EXL mice. When considered separately, the NOG-EXL mice humanized in-house were slightly less affected than the commercially available huNOG-EXL mice, but this difference was not statistically significant, and only the NOG-EXL mice humanized in-house were significantly less affected than humanized NSG-SGM3 mice (Supplemental Figure S2).
The pancreas of all humanized NSG-SGM3 mice was severely infiltrated by mast cells, resulting in expansion of the interstitium and severe loss of parenchyma (Fig. 2a). Most of the mast cells had small, indistinct cytoplasmic granules (morphology consistent with human origin), while fewer mast cells had abundant basophilic cytoplasmic granules (morphology consistent with murine origin). The identity and origin of the mast cells was confirmed by IHC for human c-KIT (Fig. 2a, inset). Some mice also had low numbers of eosinophils in the pancreas. Non-humanized NSG-SGM3 mice had a mild infiltration of murine mast cells. Humanized NOG-EXL mice did not exhibit mastocytic or eosinophilic infiltration of the pancreas (Fig. 2b).
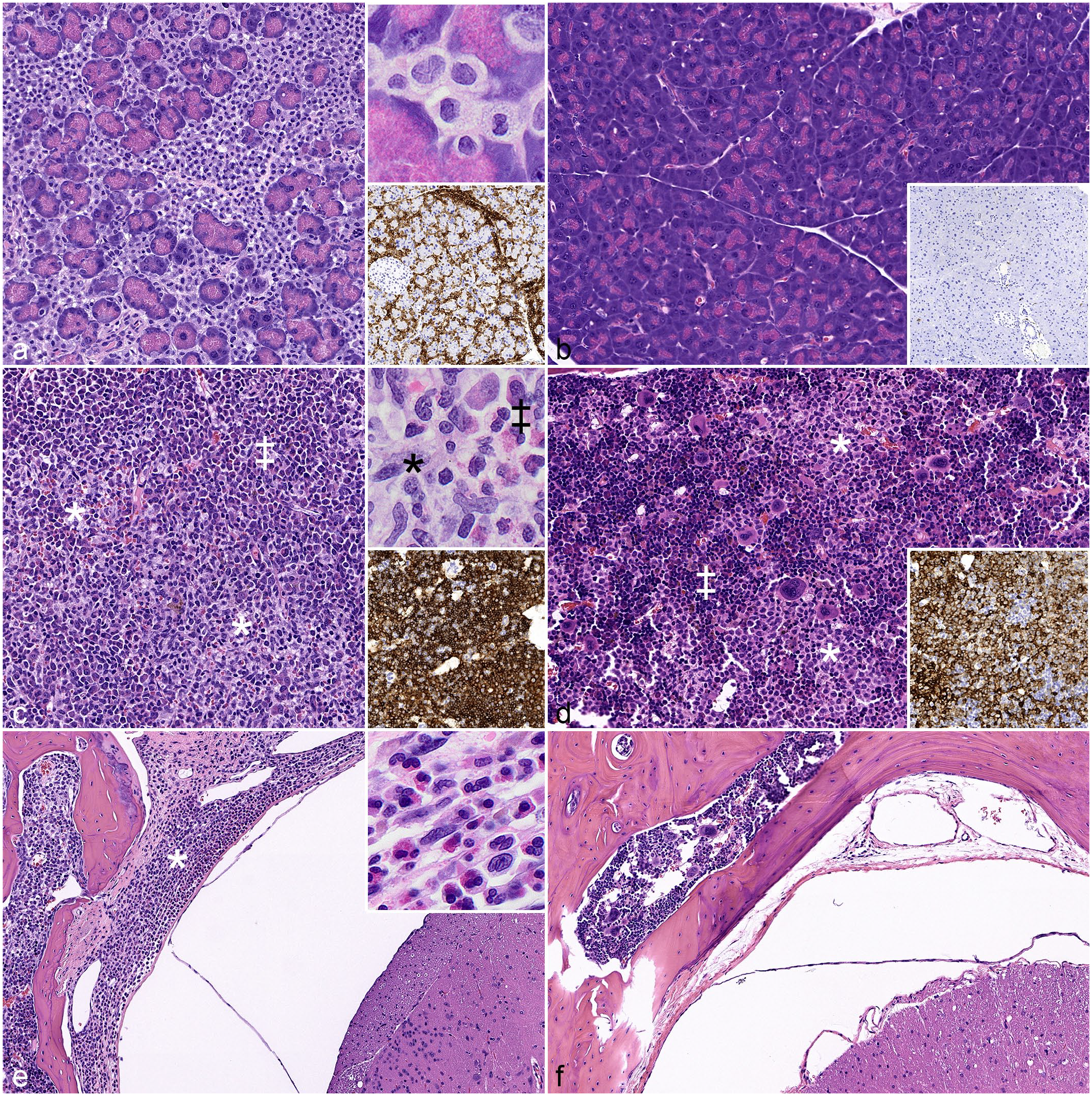
Comparison of myeloid cell hyperactivation (MCH) syndrome in the pancreas, bone marrow, and meninges. (a, c, e) Humanized NSG-SGM3 mice. (b, d, f) Commercially available huNOG-EXL mice. (a, b) Pancreas. Severe interstitial mast cell infiltration in the humanized NSG-SGM3 but not the huNOG-EXL mouse. Hematoxylin and eosin (HE). Upper inset in (a), higher magnification of infiltrating mast cells. Lower inset in (a) and (b), c-KIT immunohistochemistry (IHC). (c, d) Bone marrow. Extensive replacement by human hematopoietic cells in both strains. Increased eosinophilopoiesis (‡) and histiocytic aggregates (*) are more prominent in the humanized NSG-SGM3 mouse. HE. Upper inset in (c), higher magnification of eosinophilic precursors (‡) and histiocytes (*). Lower inset in (c) and (d), human CD45 IHC. (e, f) Spine. Infiltration of eosinophilic precursors and histiocytes into meninges of the humanized NSG-SGM3 (*) but not the huNOG-EXL mouse. HE. Inset (e), higher magnification of eosinophilic precursors and histiocytes. HE.
The bone marrow in humanized mice of both strains was populated approximately 60%–80% by human leukocytes (huCD45+), primarily of eosinophil lineage, which segmentally replaced the normal hematopoietic tissue (Fig. 2c–d). Eosinophils and eosinophilic precursors had distinct cytoplasmic granules, consistent with human morphology (Supplemental Figure S3). The human origin of these cells was confirmed by IHC for human-specific CD45 (Fig. 2c inset); the cells were negative for mouse-specific CD45. Most animals of both strains also had nodules of histiocytes in the bone marrow. In addition, in 7 of 10 humanized NSG-SGM3 mice, eosinophilic and histiocytic infiltrates extended from the bone marrow of the vertebrae into the meninges of the spinal cord (Fig. 2e). This was not observed in any humanized NOG-EXL mice (Fig. 2f).
In all mice, the splenic parenchyma lacked normal architecture (Fig. 3a, b), consistent with background immunodeficiency. Aggregates of mononuclear cells positive for human CD45 expanded periarteriolar regions, forming a poorly organized white pulp-like compartment primarily composed of lymphoid cells (Pax5+ or CD3+) that did not form distinct periarteriolar lymphoid sheaths or follicular domains. The red pulp was composed of a dispersed, approximately equal mixture of mouse and human CD45+ cells. The human cells were composed of approximately equal numbers of myeloid (CD33+) and lymphoid (Pax5+ or CD3+) cells. There were also scattered c-KIT+ human mast cells throughout the spleen, comprising approximately 10% of the total cells. In humanized NSG-SGM3 mice, histiocytes occasionally formed nodules in the red pulp, while humanized NOG-EXL mice had only few individual huCD33+ cells (Fig. 3a, b, inset). Multinucleated giant cells were present in the red pulp in all (10/10) humanized NSG-SGM3 mice (Fig. 3a) and fewer (6/22) humanized NOG-EXL mice.
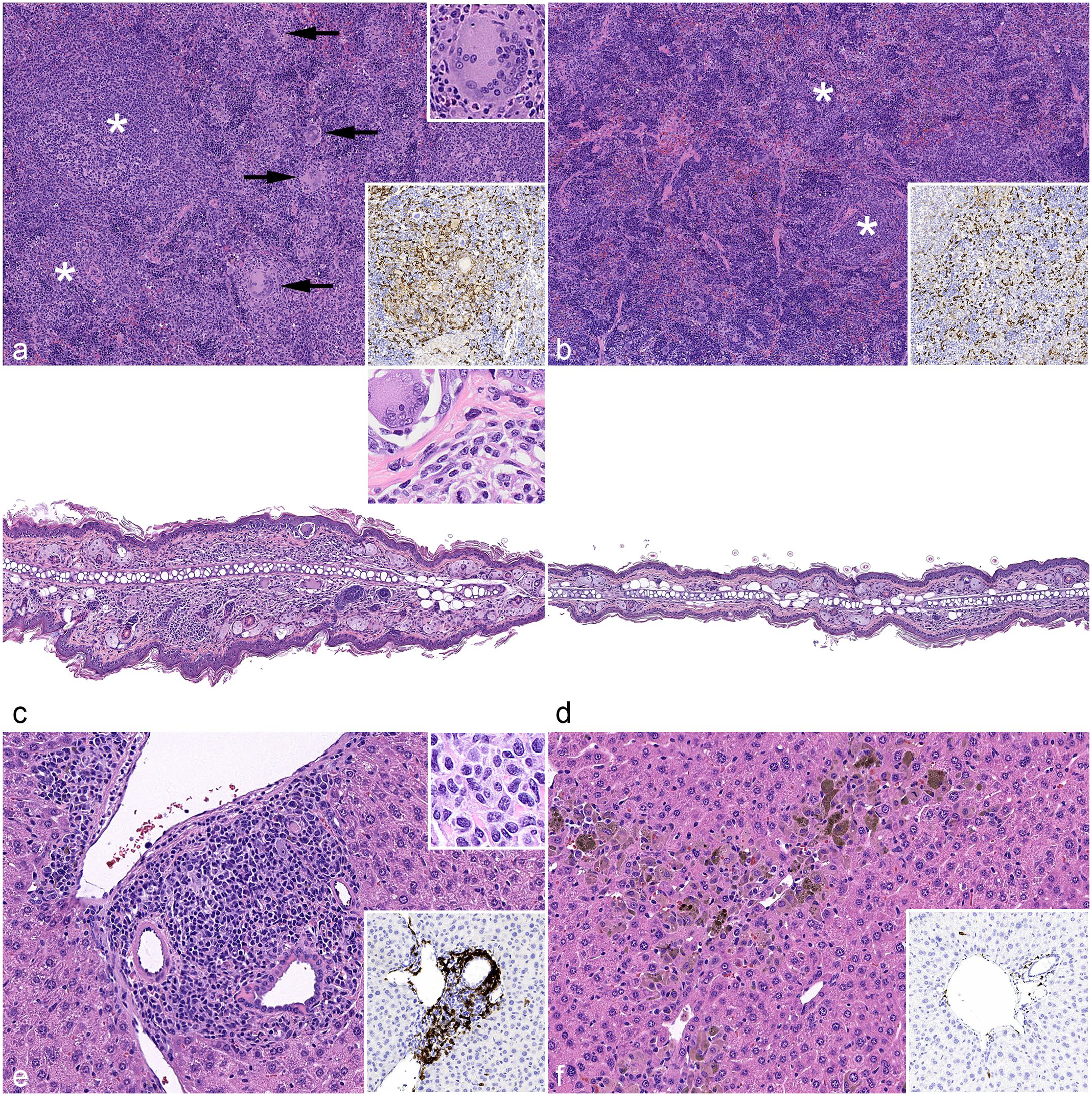
Comparison of myeloid cell hyperactivation (MCH) syndrome in the spleen, skin, and liver. (a, c, e) Humanized NSG-SGM3 mice. (b, d, f) Commercially available huNOG-EXL mice. (a, b) Spleen. Infiltration/expansion of human immune/inflammatory cells in both strains, with increased mononuclear aggregates forming vague white pulp-like structures (*). Increased histiocytes with multinucleated giant cells are present in red pulp in the humanized NSG-SGM3 mouse (panel [a], arrows and upper inset). Hematoxylin and eosin (HE). Lower inset of both panels, human CD33 immunohistochemistry (IHC). (c, d) Pinna. (c) Granulomatous interface dermatitis in a humanized NSG-SGM3 mouse. (d) Normal pinna in a huNOG-EXL mouse. Inset in (c), higher magnification of infiltrating cells. HE. (e, f) Liver. (e) Mononuclear portal infiltrates in a humanized NSG-SGM3 mouse. (f) Centrilobular to random histiocytic infiltrate with pigment in a huNOG-EXL mouse. HE. Upper inset in (e), higher magnification of infiltrating cells. HE. Lower inset in (e) and (f), c-KIT IHC.
Humanized NSG-SGM3 mice (8/10) also exhibited granulomatous dermatitis, which predominantly involved the face and ears, often destroying hair follicles and occasionally targeting the dermoepidermal junction (Fig. 3c). The majority of humanized NOG-EXL mice had no skin lesions (Fig. 3d); 2/22 humanized NOG-EXL and 1/5 non-humanized NSG-SGM3 mice had mild histiocytic, granulomatous, and/or neutrophilic dermal infiltrates.
The livers of humanized mice of both strains demonstrated a similar overall severity of lesions but differed in the character and distribution of the infiltrating cells. In humanized NSG-SGM3 mice, a mononuclear infiltrate expanded portal regions and multifocally extended into the hepatic parenchyma (Fig. 3e). Approximately 70%–80% of these cells were human myeloid cells (huCD33+), and approximately 30%–40% of those cells were mast cells. Due to the faint granulation of human mast cells, these cells were difficult to distinguish on hematoxylin and eosin staining but were demonstrated by positive c-KIT (Fig. 3e, inset) and mast cell tryptase (Supplemental Figures S4a, b) immunolabeling. The remaining myeloid cells were predominantly macrophages, which occasionally contained cytoplasmic hemosiderin (confirmed by Prussian blue staining, Supplemental Figure S4c) and exhibited mitotic activity. In some animals, low numbers of eosinophils were also present. Individual hepatocytes adjacent to the cellular infiltrate were necrotic. In both groups of humanized NOG-EXL mice, histiocytic infiltrates were scattered throughout the hepatic parenchyma and/or clustered around central veins (Fig. 3f). Similar to humanized NSG-SGM3 mice, approximately 70%–80% of these cells were huCD33+, but in humanized NOG-EXL mice, the macrophages were larger with increased cytoplasm, more frequent multinucleation, and increased frequency and amount of hemosiderin (confirmed by Prussian blue staining, Supplemental Figure S4d), as well as active erythrophagocytosis with intact red blood cells within the cytoplasm (Supplemental Figure S4e). Some humanized NOG-EXL mice exhibited individual hepatocyte necrosis, but it was less common than in humanized NSG-SGM3 mice. Individual human mast cells were scattered in the hepatic parenchyma of humanized NOG-EXL mice, comprising 5%–10% of the infiltrate (Fig. 3f, inset).
Several other organs were less commonly affected. Four out of 10 humanized NSG-SGM3 mice had histiocytic to eosinophilic infiltrates in the salivary glands; the salivary glands of humanized NOG-EXL mice were not affected. In both strains of mice, the lungs occasionally contained minimal to mild histiocytic to granulomatous infiltrates. A severe transmural eosinophilic and mastocytic gastritis was observed in one humanized NSG-SGM3 mouse. In humanized NOG-EXL mice, tumor engraftment was not associated with significant differences in the composition, distribution, and severity of the described lesions (Supplemental Figure S5).
Hematology
Despite a much longer post-humanization interval in NOG-EXL mice (i.e., 22 weeks) compared to NSG-SGM3 mice (i.e., 16 weeks), both strains exhibited similar levels of anemia, with a mean hemoglobin concentration of 8.0 (standard deviation 2.9) g/dL in humanized NSG-SGM3 and 6.4 (±1.9) g/dL in humanized NOG-EXL mice. Reference hemoglobin concentrations were measured in non-humanized groups with a mean of 13.7 (±0.4) and 13.2 (±0.6) g/dL in NSG-SGM3 and NOG-EXL mice, respectively (Fig. 4a). The humanized NSG-SGM3 mice had significantly increased total leukocyte counts at the time of death compared to non-humanized NSG-SGM3 mice and humanized NOG-EXL mice (Fig. 4b). When considered separately, the NOG-EXL mice humanized in-house and the commercially available huNOG-EXL mice had similar hematologic findings (Supplemental Figure S6). Furthermore, only the NOG-EXL mice humanized in-house had significantly lower total leukocyte counts than the humanized NSG-SGM3 mice (Supplemental Figure S5).
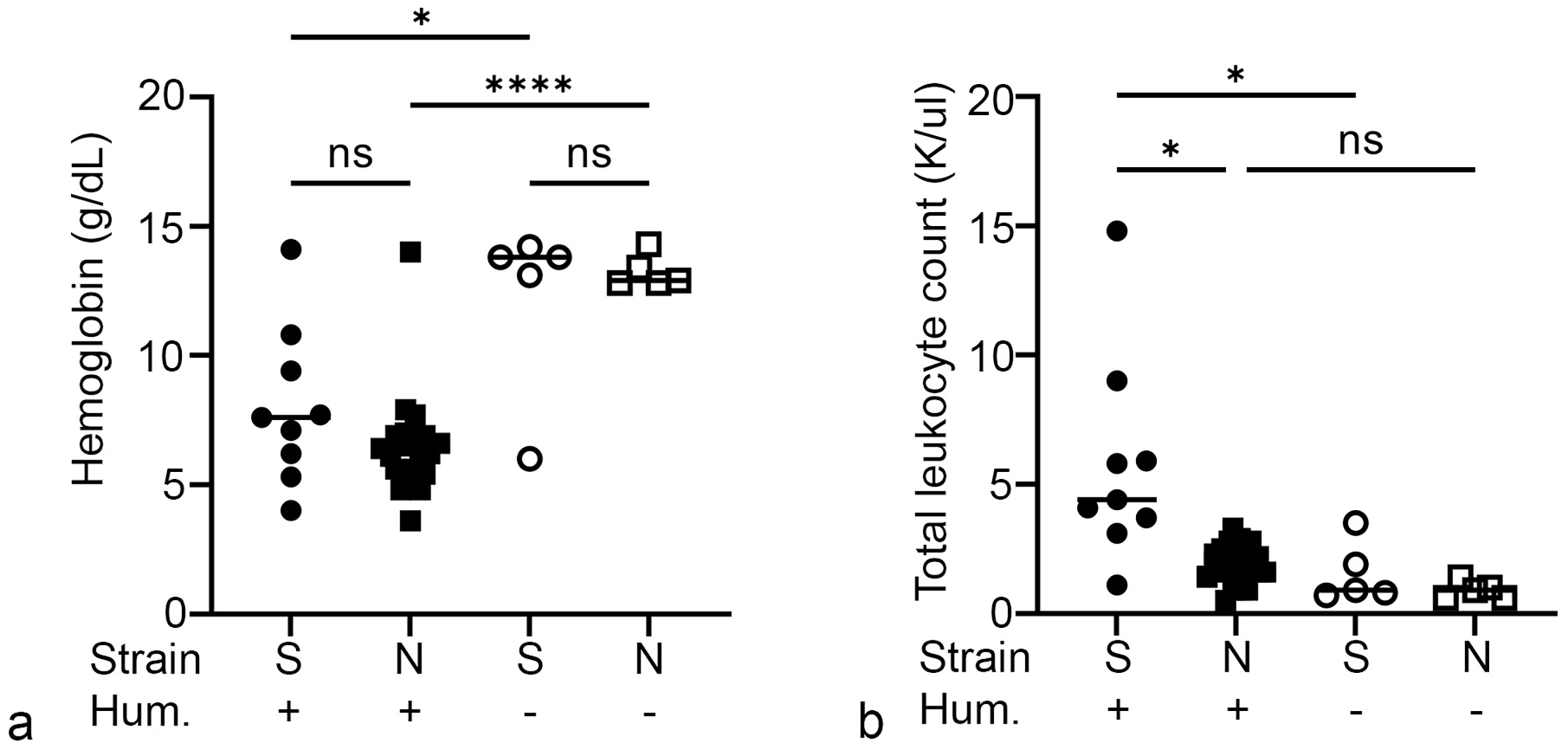
Comparison of hematologic parameters. (a) Anemia is similarly severe in both strains of humanized mice. (b) Humanized NSG-SGM3 mice have elevated leukocyte counts. S, NSG-SGM3; N, NOG-EXL; Hum., humanized. One-way analysis of variance (ANOVA) with Tukey’s test. ns, not significant. *P < .05, ****P < .0001. n = 22 mice/group (humanized NOG-EXL), 9 mice/group (humanized NSG-SGM3), and 5 mice/group (non-humanized).
Assessment of Lymphoid Repopulation From Engrafted Human HSC
Engraftment of human lymphocytes was assessed by IHC and flow cytometry. In both strains, human CD45+ cells comprised 80%–90% of the lymph node populations, with approximately 40%–60% T cells and 25%–40% B cells distributed throughout the node lacking the typical compartmentalization of the normal architecture (Supplemental Figures S7a–d). The spleens of both strains of mice had a dispersed population of human B and T cells throughout the red pulp, comprising 15%–30% of the cells. In both strains, these cells also formed aggregates, often centered around blood vessels (Fig. 3a, b). These aggregates contained approximately equal proportions of B cells between the two strains and slightly more T cells in humanized NSG-SGM3 mice. B and T cells were randomly distributed in these aggregates, without forming distinct periarteriolar lymphoid sheaths or follicular domains. Although the bone marrow was primarily replaced by human myeloid cells, 10%–30% of the marrow cells in both strains were B cells, and 5%–10% of the cells in humanized NOG-EXL and 10%–20% of the cells in humanized NSG-SGM3 were T cells. In the liver, humanized NOG-EXL mice had only scattered B and T cells, while humanized NSG-SGM3 mice had substantial T cells (20%–50%) and B cells (5%–20%) in the infiltrate (Supplemental Figures S7e–h).
The engraftment in peripheral blood was assessed by flow cytometry at 10 weeks after humanization for the NSG-SGM3 and NOG-EXL mice humanized in-house (Supplemental Figure S8). Humanized NSG-SGM3 mice had an average of 15% (159 cells/µl peripheral blood) human lymphoid (CD3+ or CD19+) cells and 35% (323 cells/µl) human myeloid (CD33+) cells in peripheral blood, while humanized NOG-EXL mice had averages of 11% (97 cells/µl) and 4% (34 cells/µl), respectively.
Discussion
In this study, we compared the lesions associated with transplantation of human CD34+ HSCs in 2 similar immunodeficient mouse strains that express human myeloid stimulatory cytokines to improve human myeloid cell engraftment. While both strains develop lesions characterized by unregulated expansion and activation of human myeloid cells and consistent with MCH syndrome, the NOG-EXL mice survived longer, enabling the study endpoint to be reached. Overall, the humanized NOG-EXL mice had less severe lesions with limited organ/tissue distribution. These differences appear even more significant considering the much longer post-humanization interval endured by NOG-EXL mice (22 weeks) than NSG-SGM3 mice (16 weeks). Importantly, humanized NOG-EXL mice lacked the prominent expansion and infiltration of human mast cells. On the contrary, the pancreas in the humanized NSG-SGM3 mice was invariably infiltrated and replaced by numerous human and fewer mouse mast cells, as previously reported.10,23 Interestingly, mast cells were present in significant numbers in the liver as well in humanized NSG-SGM3 mice. The differences in mast cell infiltration are likely due to the transgenic expression of the human KIT ligand (also known as stem cell factor or Steel factor) in NSG-SGM3 mice and lack thereof in NOG-EXL mice. While expression of KIT ligand improves engraftment of human hematopoietic cells,22,24 this cytokine and its c-KIT tyrosine kinase receptor also play an essential role in mast cell development and function. 24 Considering the severe lesions affecting the pancreas and, to a lesser degree, the liver, the massive expansion and activation of human mast cells, which is presumed to result from unmodulated c-KIT stimulation, likely plays a significant role in exacerbating the MCH phenotype and determining the severe morbidity and mortality in humanized NSG-SGM3 mice. Further supporting this evidence, Takagi et al 22 showed that NSG mice expressing membrane-bound KIT ligand under a ubiquitous promoter became moribund as early as 8 weeks after human hematopoietic cell transplantation, exhibiting anemia and increased mast cells in the spleen and bone marrow; however, other organs were not examined.
Interestingly, the hepatic lesions in humanized NOG-EXL mice were similar in severity to humanized NSG-SGM3 mice but differed in quality. The portal distribution of the immune cell infiltrates and the greater frequency of hepatocellular necrosis in humanized NSG-SGM3 mice likely led to more severe impairment of liver function than the multifocal random to centrilobular distribution and less-frequent hepatocellular necrosis observed in the humanized NOG-EXL mice. In addition, the human myeloid infiltrates in the livers of humanized NSG-SGM3 mice in this study included a relatively large proportion of mast cells, which has not been reported in the liver in previous descriptions of this disorder.10,23 The increased erythrophagocytosis, especially given the presence of intact erythrocytes, in the liver of humanized NOG-EXL mice, may reflect slower progression and decreased chronicity of the lesions compared to those observed in the humanized NSG-SGM3 mice. More acute lesions at a longer post-humanization interval (22 vs 16 weeks) underscores the delayed clinical course of the humanized NOG-EXL mice. Monitoring of liver function (eg, by chemistry profiles) may represent a good indicator of the progression of the MCH phenotype.
HLH occurs as a familial or acquired disease in humans and is associated with genetic loss of cytotoxic function in immune cells or secondary to infectious or neoplastic conditions, especially Epstein-Barr virus infection. HLH is characterized by dysregulation of activated cytotoxic T cells, natural killer cells, and macrophages. 17 MAS is considered a subset of HLH that mainly occurs as a complication of various chronic rheumatic diseases of childhood. 17 Several studies have described MAS or HLH-like lesions in humanized NSG-SGM3 mice or the similar NRG-SGM3 strain,10,23,28,30 as well as in NOG mice. 21 The NSG-SGM3 or NRG-SGM3 mice developed these syndromes after humanization with CD34+ HSCs derived from umbilical cord blood, fetal liver, or bone marrow from oncologic patients.10,23,28,30 Cases associated with patient-derived tumor xenograft transplantation have also been reported. 10 The NOG mice described by Sato et al 21 were humanized with CD34+ HSCs but did not develop HLH-like lesions until after infection with Epstein-Barr virus. Humanized NSG mice have been reported to develop histiocytic proliferation, but histiocytes lacked the hyperactivated phenotype of MAS/HLH.2,10 The MAS observed in humanized NSG-SGM3 and NOG-EXL mice is attributed to the supraphysiologic expression of human GM-CSF and, to a lesser extent, IL-3.4 The less severe phenotype in the NOG-EXL mice is likely due to remarkable differences in expression levels, as the cytokines are driven by different viral promoters/enhancers in NSG-SGM3 and NOG-EXL mice.1,7 In this context, Maser et al 13 detected 400-fold higher GM-CSF serum levels in humanized NSG-SGM3 mice than humanized NOG-EXL mice. Significantly increased levels of other proinflammatory signals were also detected in humanized NSG-SGM3 mice including human interferon-γ, IL-6, IL-8, monocyte chemoattractant protein (MCP)-1, and tumor necrosis factor (TNF)-α. 13
In this study, we measured the presence of human leukocytes by flow cytometry in peripheral blood as well as by IHC in tissues. In peripheral blood, the percentage of human lymphocytes was similar between NSG-SGM3 and NOG-EXL mice humanized in-house. However, the percentage of myeloid cells in the peripheral blood of humanized NOG-EXL mice was significantly lower than that of humanized NSG-SGM3 mice, even though histiocytic infiltrates were present in the tissues. This difference may be due to the early timepoint of flow cytometry analysis (10 weeks after humanization for flow cytometry versus 16 and 22 weeks after humanization for tissue collection in NSG-SGM3 and NOG-EXL, respectively) or a poor correlation between repopulation of peripheral blood and tissues as observed in previous studies.2,23 A limitation of this study is the lack of flow cytometric analysis at the time of death; however, the IHC analysis shows that engraftment of human lymphocytes in lymphoid tissues was similar between the two strains.
The spleen, bone marrow, and liver had mildly to moderately increased numbers of T cells in humanized NSG-SGM3 mice compared to humanized NOG-EXL mice. A similar trend was observed for B cells in the liver. It is unclear to what extent these lymphocytes, in particular T cells, contribute to the lesions described here. Interestingly, previous descriptions of MAS specifically report a very limited involvement of lymphocytes. 23 However, in humans and other mouse models, cytotoxic T cells are key mediators of HLH. It is therefore possible that the T-cell infiltrates may contribute to the more severe phenotype observed in the humanized NSG-SGM3 mice. There may also be a component of graft versus host disease in these mice that cannot be clearly separated from the MCH syndrome. 3 The interface dermatitis we observed in the skin of a subset of humanized NSG-SGM3 mice may also be an example of graft versus host disease, although, interestingly, the dermal infiltrates were primarily histiocytic to granulomatous rather than lymphocytic.
Our work compared an off-the-shelf commercially available product generated via a proprietary humanization protocol (huNOG-EXL mice from Taconic Biosciences) with NOG-EXL and NSG-SGM3 mice humanized according to a protocol developed in our facility at the University of Pennsylvania. In this context, one of the major limitations of this study is the assessment of models where the overall humanization procedures differed in terms of preconditioning, source and amount of human CD34+ HSCs delivered, as well as route of engraftment. While recognizing the importance of these experimental variables, we believe that the results of our comparison remain relevant considering that none of these different approaches have shown a major impact on the latency and severity of the phenotype. Evidence from this study and previous experiments clearly indicates that NSG-SGM3 mice humanized under a variety of conditions invariably develop a severe and frequently lethal MCH phenotype that manifested before 22 weeks after humanization and as early as 4 weeks in some mice.10,23,28,30 In addition, we observed clinically negligible phenotype variations when comparing the huNOG-EXL product from Taconic Biosciences and the NOG-EXL cohort humanized in our facility. Importantly, these results indicate that, despite the different protocols used for humanization, the NOG-EXL model is clinically very stable over a much longer post-humanization interval, ensuring reproducible results across different experimental settings.
In summary, we compared the severity and features of post-transplant phenotype in two humanized mouse strains (NSG-SGM3 and NOG-EXL). To the authors’ knowledge, this is the first description of MCH syndrome in humanized NOG-EXL mice. While both strains developed lesions, humanized NOG-EXL mice exhibited less severe MAS and eosinophil hyperplasia and did not develop mast cell proliferation, thus allowing the achievement of the experimental endpoint in a patient-derived xenograft glioblastoma organoid model. Although the development of these post-transplant disorders remains a hindrance to very long-term studies, the humanized NOG-EXL model nevertheless generates a longer window of opportunity for studies using myeloid engraftment-enhancing mouse models.
Supplemental Material
sj-pdf-1-vet-10.1177_03009858231222216 – Supplemental material for Humanization with CD34-positive hematopoietic stem cells in NOG-EXL mice results in improved long-term survival and less severe myeloid cell hyperactivation phenotype relative to NSG-SGM3 mice
Supplemental material, sj-pdf-1-vet-10.1177_03009858231222216 for Humanization with CD34-positive hematopoietic stem cells in NOG-EXL mice results in improved long-term survival and less severe myeloid cell hyperactivation phenotype relative to NSG-SGM3 mice by Elinor Willis, Jillian Verrelle, Esha Banerjee, Charles-Antoine Assenmacher, James C. Tarrant, Nicholas Skuli, Moriah L. Jacobson, Donald M. O’Rouke, Zev A. Binder and Enrico Radaelli in Veterinary Pathology
Supplemental Material
sj-xlsx-2-vet-10.1177_03009858231222216 – Supplemental material for Humanization with CD34-positive hematopoietic stem cells in NOG-EXL mice results in improved long-term survival and less severe myeloid cell hyperactivation phenotype relative to NSG-SGM3 mice
Supplemental material, sj-xlsx-2-vet-10.1177_03009858231222216 for Humanization with CD34-positive hematopoietic stem cells in NOG-EXL mice results in improved long-term survival and less severe myeloid cell hyperactivation phenotype relative to NSG-SGM3 mice by Elinor Willis, Jillian Verrelle, Esha Banerjee, Charles-Antoine Assenmacher, James C. Tarrant, Nicholas Skuli, Moriah L. Jacobson, Donald M. O’Rouke, Zev A. Binder and Enrico Radaelli in Veterinary Pathology
Footnotes
Acknowledgements
The authors would like to thank Megan McBride (Taconic Biosciences) for the insightful comments.
Supplemental material for this article is available online.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Moriah L. Jacobson is an employee of Taconic Biosciences Inc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the GBM Translational Center of Excellence of the Abramson Cancer Center at University of Pennsylvania. The authors affiliated with the Penn Vet Comparative Pathology Core are partially subsidized by the Abramson Cancer Center Support Grant (P30 CA016520) and the Penn Vet Institute for Infectious & Zoonotic Diseases; the Aperio Versa 200 scanner used for imaging was acquired through an NIH Shared Instrumentation Grant (S10 OD023465-01A1); the Leica BOND RXm instrument used for IHC was acquired through the Penn Vet Institute for Infectious & Zoonotic Diseases Core pilot grant opportunity 2022.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.