
Abstract
Canavan disease is an autosomal recessive leukodystrophy caused by mutations in the gene encoding aspartoacylase (ASPA), which hydrolyses N-acetylaspartate (NAA) to acetate and aspartate. A similar feline neurodegenerative disease associated with a mutation in the ASPA gene is reported herein. Comprehensive clinical, genetic, and pathological analyses were performed on 4 affected cats. Gait disturbance and head tremors initially appeared at 1 to 19 months of age. These cats eventually exhibited dysstasia and seizures and died at 7 to 53 months of age. Magnetic resonance imaging of the brain revealed diffuse symmetrical intensity change of the cerebral cortex, brainstem, and cerebellum. Gas chromatography–mass spectrometry analysis of urine showed significant excretion of NAA. Genetic analysis of the 4 affected cats identified a missense mutation (c.859G>C) in exon 6 of the ASPA gene, which was not detected in 4 neurologically intact cats examined as controls. Postmortem analysis revealed vacuolar changes predominantly distributed in the gray matter of the cerebrum and brain stem as well as in the cerebellar Purkinje cell layer. Immunohistochemically, these vacuoles were surrounded by neurofilaments and sometimes contained MBP- and Olig2-positive cells. Ultrastructurally, a large number of intracytoplasmic vacuoles containing mitochondria and electron-dense granules were detected in the cerebral cortex. All 4 cats were diagnosed as spongy encephalopathy with a mutation in the ASPA gene, a syndrome analogous to human Canavan disease. The histopathological findings suggest that feline ASPA deficiency induces intracytoplasmic edema in neurons and oligodendrocytes, resulting in spongy degeneration of the central nervous system.
Keywords
Canavan disease (CD) is an autosomal recessive progressive neurodegenerative disease characterized by macrocephaly and spongy degeneration of the cerebral white matter. 1 Clinical signs of this disease generally appear during the first few months after birth, manifest as poor head control and marked developmental delay, and progress to macrocephaly, optic atrophy, seizures, and hypertonia with death in early childhood. 13 Human CD is caused by loss-of-function mutations in the ASPA gene encoding aspartoacylase (ASPA). This enzyme is expressed in oligodendroglial cells and converts N-acetylaspartate (NAA), one of the most abundant amino acids in the mammalian brain, into acetate and aspartic acid. The concentration of NAA is elevated in the CSF (cerebrospinal fluid), urine, and blood of affected individuals. 17
In the brain, ASPA is physiologically present in oligodendrocyte progenitor cells and oligodendrocytes, while smaller amounts are present in microglia and brainstem neurons, 21 and its substrate NAA is synthesized in neuronal mitochondria. Although NAA has multiple functions in the brain, the supply of acetate via the hydrolysis of NAA in oligodendrocytes is critically involved in the synthesis of myelin lipids. 4 A lack of acetate in oligodendrocytes due to ASPA dysfunction may play an important role in the pathogenesis of human CD. However, not all histopathological hallmarks of CD may be explained by impaired myelin synthesis and maintenance, and the mechanistic link between the observed brain lesions and absence of ASPA remains unclear. The diagnosis of human CD is based on functional, genotypic, and phenotypic examinations. In addition to these analyses, detecting the aberrant excretion of NAA in urine and magnetic resonance imaging (MRI) examinations of the brain are the least invasive methods for confirming human CD. 17
In animals, there are several transgenic mouse models of CD that carry null alleles of the ASPA gene, such as ASPAnur7, 7,23 ASPAlacZ/lacZ, 20 ASPATm1Mata, 18 and ASPAdeaf14 mice. 6 Histologically, all of these mice develop similar spongy degeneration throughout the central nervous system. Furthermore, several types of leukodystrophy have been described in cattle, 9 sheep, 12 cats, 15 headgehogs, 8 silver foxes, 11 and dogs. 16,24,25 The majority of these cases are considered to be inherited diseases. The most common clinical presentation is progressive neurological dysfunction beginning in the neonatal or juvenile period. 15,16,24,25 Major histological lesions are vacuolar changes or spongiosis of the white matter of the brain and spinal cord. However, a spontaneous genetic mutation in the ASPA gene has never been reported in domestic animals.
We herein describe the clinical, genetic, and pathological features in 4 mixed-breed cats with the same missense mutation in the ASPA gene that developed a progressive neurological disease beginning in the first few months after birth.
Materials and Methods
Animals
Four female mixed-breed cats exhibiting neurological signs consistent with intracranial disease were examined (Table 1). These cats were bred in different regions in Japan and considered to be an unrelated pedigree. Genetic and biochemical analyses were performed at Kagoshima University. MRI (cases 1–4) and gas chromatography–mass spectrometry (GC-MS) analyses of urine (case 2) were performed within 1 year of clinical manifestations. After the spontaneous death of all 4 cats, necropsies were performed on the day of the death, and tissue samples including brain, were collected for histopathology, immunohistochemistry, and electron microscopy.
Cases Examined in the Present Study.
ASPA Gene Analysis
In the molecular analysis of the ASPA gene, genomic DNA was extracted from frozen brain tissue using the DNeasy Blood & Tissue Kit (Qiagen). Samples from 4 neurologically intact female mixed-breed cats were also examined for the gene analysis as control cases. Purified DNA samples were used for subsequent PCR (polymerase chain reaction). The feline ASPA gene-specific PCR primer pair that amplifies a 223-bp fragment incorporating exon 6 of the ASPA gene was used: 5′-CAAGACTGGAAACCGCTGC-3′ (forward) and 5′-TGTGTCACAGGTGGGCTAGA-3′ (reverse). PCR was performed as follows: 30 cycles at 98 °C for 10 seconds and 60 °C for 30 seconds. PCR products were submitted for a sequence analysis (FASMAC). PCR was performed in duplicate. In silico analysis of mutations was performed using the sorting intolerant from tolerant (SIFT) algorithm.
Pathology
Tissue samples were fixed in 10% neutral-buffered formalin and embedded in paraffin wax. Formalin-fixed and paraffin-embedded tissues were sectioned at a thickness of 4 µm and then stained with hematoxylin and eosin (HE), Luxol fast blue (LFB), periodic acid-Schiff (PAS), and Alcian blue (pH 4.1).
For immunohistochemistry, consecutive sections were labeled using an immunoenzyme technique. After deparaffinization and rehydration, antigen retrieval was performed via heating. To deactivate endogenous peroxidase, sections were immersed in 1% hydrogen peroxide in methanol for 5 minutes. To avoid nonspecific binding of the antibody, sections were immersed in 8% skim milk in Tris-buffered saline. The following primary antibodies were used: mouse anti-neurofilament (clone NP1, 1:200, Millipore), rabbit anti-MBP (1:2500, DAKO), rabbit anti-GFAP (1:1000, DAKO), rabbit anti-Iba-1 (1:500, WAKO), rabbit anti-ubiquitin (1:400, Cell Signaling Technology), and rabbit anti-Olig2 (1:500, Millipore). After incubation with each primary antibody at 4 °C overnight, immunolabeled antigens were visualized using the Dako EnVision+ System (Dako) with 0.02% 3′3-diaminobenzidine plus 0.01% hydrogen peroxide as a chromogen.
For electron microscopy, formalin-fixed brain samples were washed with phosphate-buffered saline (pH 7.4) and post-fixed in 1% osmium tetroxide. Samples were dehydrated and embedded in resin Luveak-812 (Nacalai Tesque). Uranyl acetate and lead nitrate–stained ultrathin sections were examined with a transmission electron microscope (JEM-1400Plus, JEOL).
Results
Clinical Manifestation
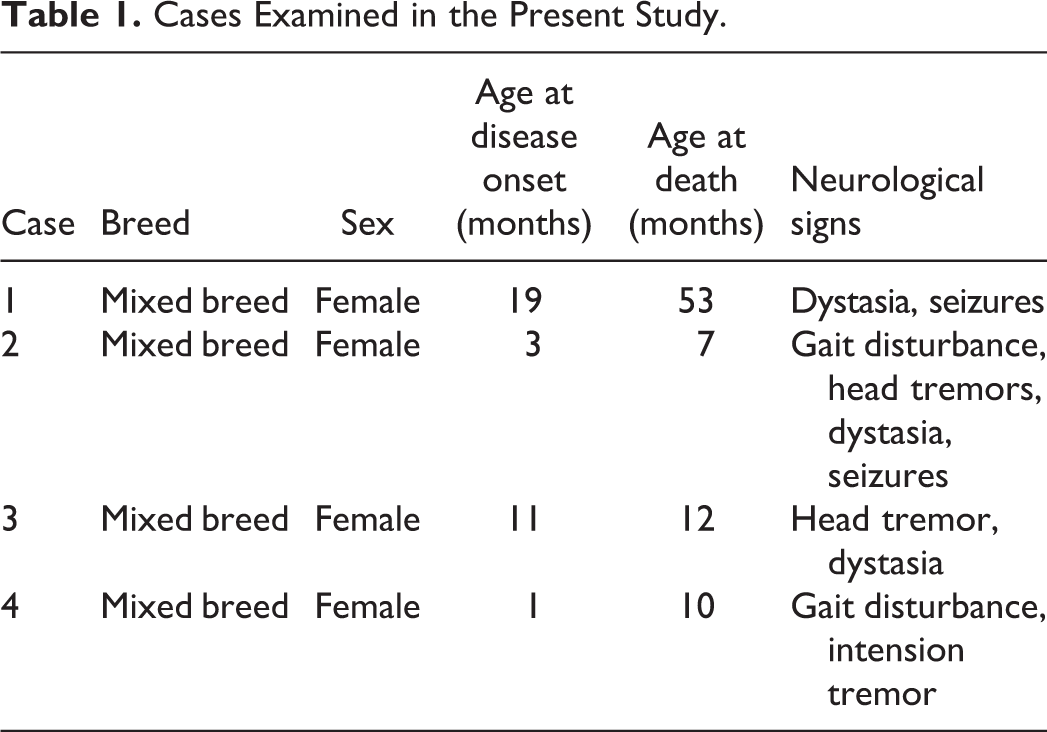
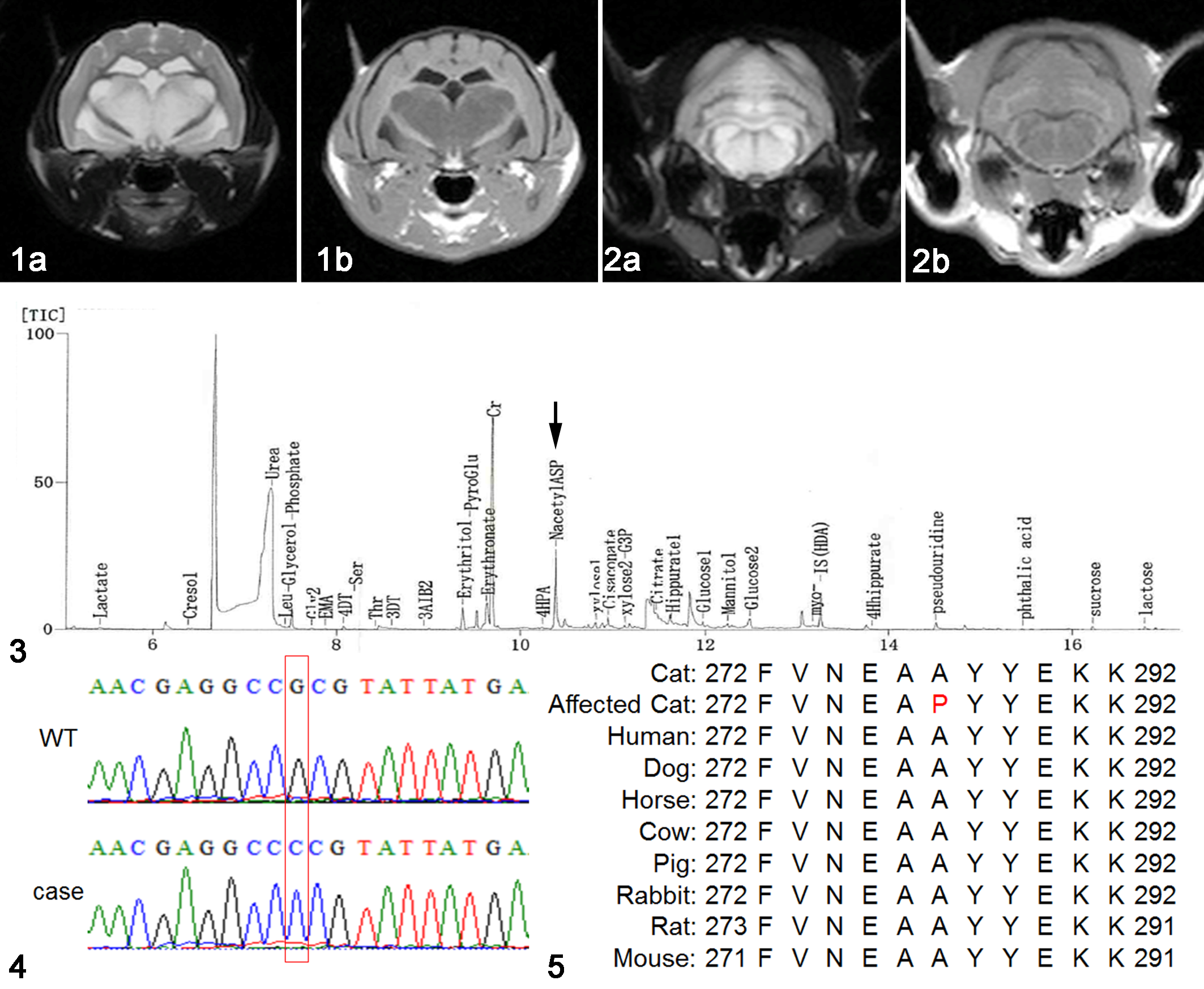
The initial clinical signs were gait disturbance and head tremors appearing between 1 and 19 months of age. Dysstasia, intension tremors, and seizures were present in the terminal stages, and the median age of death was 11 months, ranging between 7 and 53 months (Table 1). The median survival time from the onset of clinical signs was 6.5 months, ranging between 4 and 34 months. In MRI analyses, hypointense lesions on T1-weighted (T1W) images and hyperintense lesions on T2W images were detected in the cerebral cortex, hippocampus, cerebellum, and brain stem (Figs. 1, 2). The GC-MS analysis identified the aberrant excretion of NAA in the urine of case 2 (Fig. 3).
Cat.
Genetic Analysis of the ASPA Gene
A single base substitution of guanine for cytosine (c.859G>C) in exon 6 was identified in all affected cats (Fig. 4). This missense mutation is predicted to result in an amino acid substitution of a conserved alanine to proline (A287P). Multiple sequence alignments of the mutant ASPA protein with wild-type feline, murine, human, and domestic animal ASPA proteins showed that the primary sequence was highly conserved among many mammalian orthologs (Fig. 5). The possible effects of the genetic mutation on the functions of ASPA were evaluated by SIFT. The mutation showed a SIFT score of 0.00 and was predicted to be highly damaging to the functions of the ASPA protein.
Histopathological Findings
Affected cats exhibited a subtle distinction between deep cortex and subcortical white matter in the cerebrum (Fig. 6), as well as cerebral cortical atrophy and ventriculomegaly (Fig. 7). Histopathologically, severe vacuolar changes were present in the neuropil of cerebral cortex, particularly in the second to fourth cortical layers in all affected cats, and these lesions extended to the deeper cortical layers in 3 out of 4 cats (cases 2–4, Fig. 8). Vacuolar changes were also present in Ammon’s horn and the dentate gyrus of the hippocampus in all cats (Fig. 9). Vacuoles varied in size, ranging between 20 and 100 µm in diameter, and shape, and were negative with PAS, LFB, and Alcian blue stains. Irregularly large empty spaces, referred to as polycavitation, which presumably formed due to the coalescence of ruptured vacuoles, were occasionally present. Activated astrocytes characterized by an enlarged pale nucleus and activated microglial cells were scattered within the lesions of the cerebral cortex. Vacuolar changes were scarce in the subcortical white matter of the cerebrum but were associated with severe loss of axons and myelin sheaths. All affected cats, particularly case 1, had severe diffuse or perivascular astrogliosis in the cerebral white matter, and lesions were characterized by hypertrophic astrocytes with abundant eosinophilic cytoplasm (gemistocytic astrocytes). On LFB-stained sections, myelin loss was directly proportional to the degree of cortical vacuolization (Fig. 10). In the deeper white matter, myelin sheaths were not as severely affected and moderate astrogliosis was present.
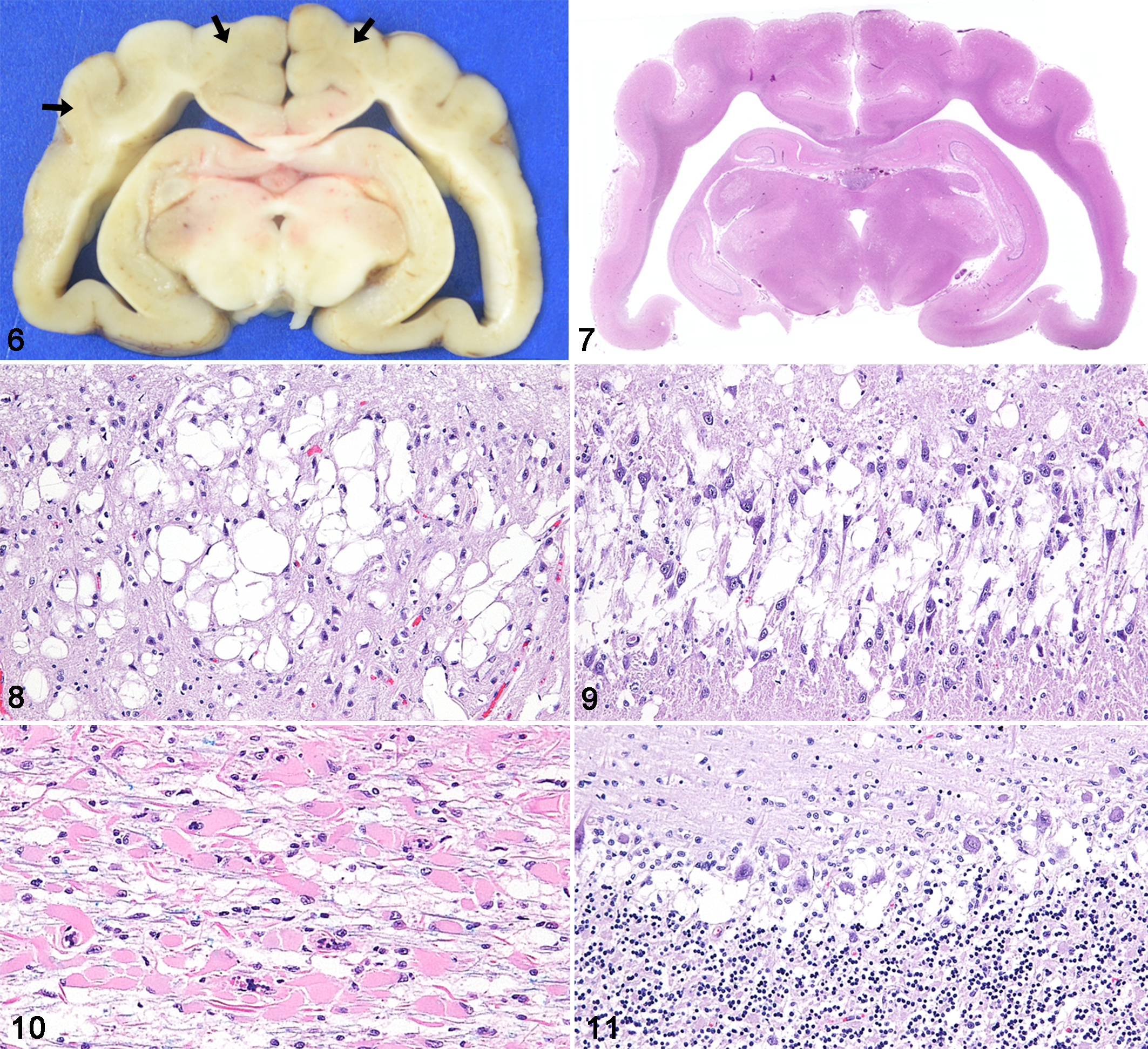
Spongy encephalopathy, brain, cat, case 1.
In the cerebellar cortex, marked vacuolar changes of the Purkinje layer as well as moderate diffuse astrogliosis were present in all affected cats (Fig. 11). There were also moderate vacuolar changes in the molecular and granular layers of 2 affected cats (cases 2 and 3). Multiple vacuolar changes with reactive astrogliosis and infiltration of activated microglial cells were present in the subcortical cerebellar white matter, while axonal degeneration and myelin loss were less prominent. In all cats, the deep cerebellar white matter was not as severely affected.
Severe vacuolar changes similar to those seen in the cerebral cortex were present in the gray matter of the midbrain, pons, and medulla oblongata of 3 cats (cases 1–3). Vacuolar changes were also detected in the white matter of the mesencephalon, pons and medulla oblongata in 2 cats (cases 2 and 3). No significant vacuolar changes were detected in the brain stem of case 4. The degree and distribution of astrogliosis and microglial infiltration corresponded to those of vacuolar changes.
The spinal cords of 2 affected cats (cases 1 and 2) and the visceral organs of one of these cats (case 2) were examined by histopathology. In the spinal cord of case 2, moderate vacuolar changes with mild astrogliosis were detected in the gray matter, while no significant lesions were present in the white matter. Furthermore, there were no significant lesions in the spinal cord of case 1 or in the peripheral nerves and visceral organs of case 2.
Immunohistochemistry and Electron Microscopy
By immunohistochemistry, the periphery of the lesional vacuoles in the cerebral cortex were intensely positive for neurofilament (Fig. 12a), but were negative for MBP, GFAP, Iba-1, and ubiquitin (Fig. 12b–e). Cells in or around the lesional vacuoles showed cytoplasmic immunoreactivity for MBP (Fig. 12b) and nuclear immunoreactivity for Olig2 (Fig. 12f). Considering both the immunohistochemical and morphological findings, these cells were considered to be oligodendrocytes. Gemistocytic astrocytes (GFAP-positive) and reactive microglial cells (Iba-1-positive) were present within cerebral white matter lesions characterized by the severe loss of myelin sheaths (Fig. 13a and b).
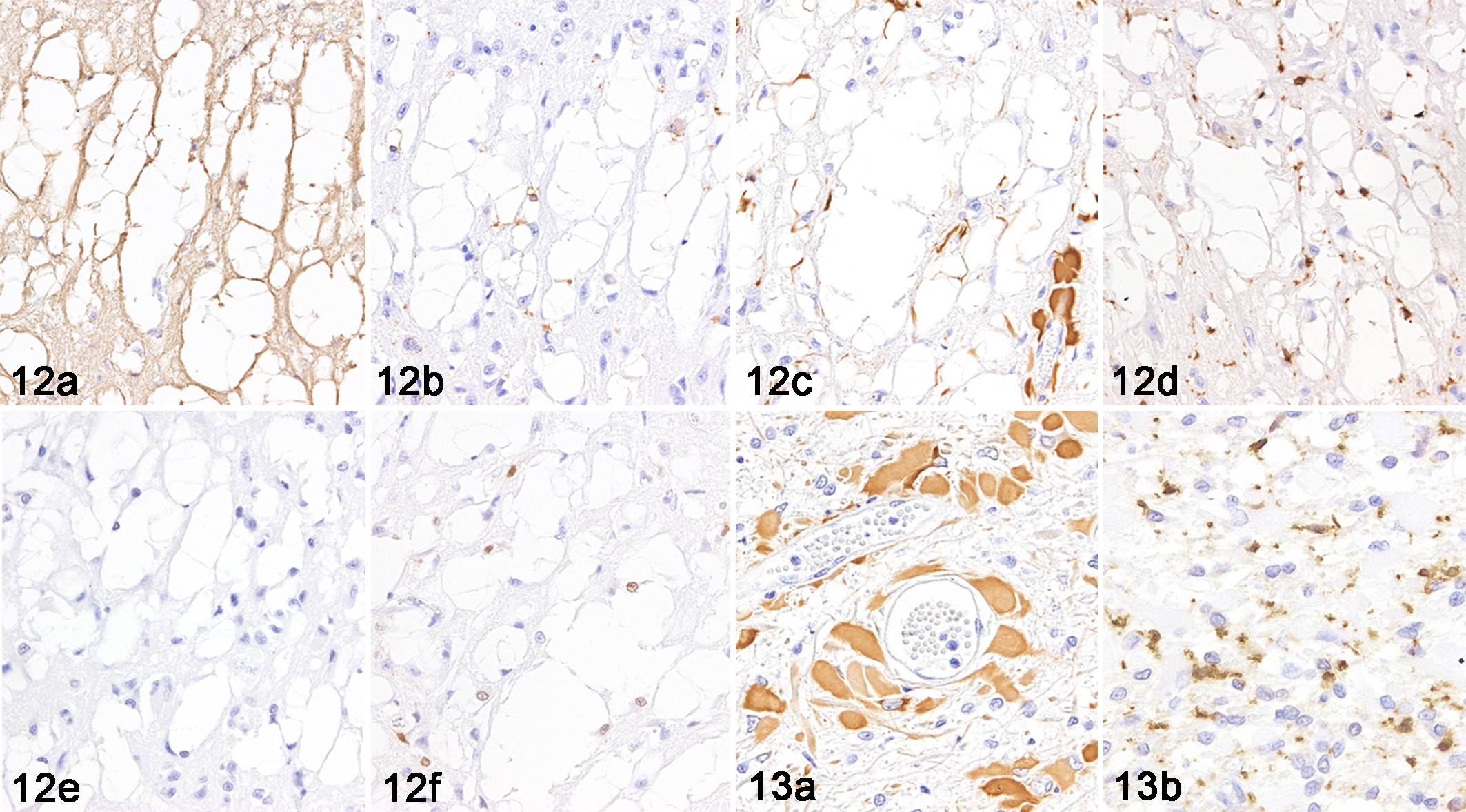
Spongy encephalopathy, cat, case 1.
Electron microscopy confirmed the presence of a large number of small vacuoles in the cerebral cortex. These vacuoles contained mitochondria and/or electron-dense granules (Fig. 14). Larger vacuoles were formed by the coalescence of ruptured vacuoles. Interspersed areas of intramyelinic vacuolization caused by the separation of lamellae at intraperiod lines between major dense lines (ie, myelin splitting) were occasionally present in the cerebral cortex (Fig. 15).
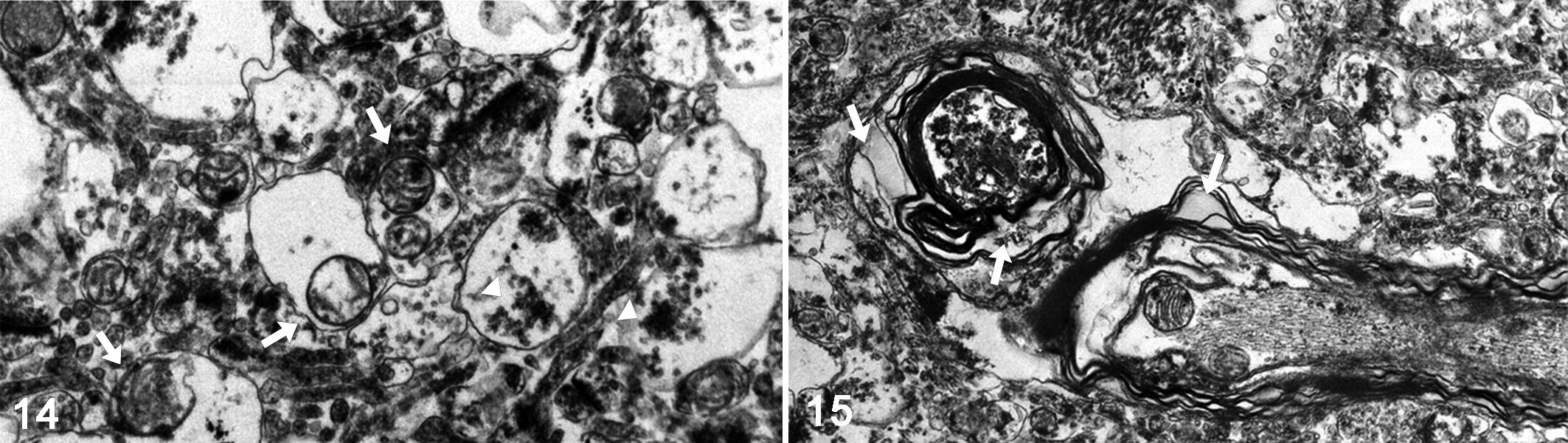
Spongy encephalopathy, cat, case 1, cerebral cortex. Transmission electron microscopy.
Discussion
Human CD is a rare vacuolating leukodystrophy that is caused by ASPA deficiency. ASPA deficiency leads to the accumulation of NAA, which results in spongy degeneration of white matter in the brain. 1 Spongy leukodystrophy has been reported in domestic animals, and its pathological features were similar to human CD; however, a genetic analysis has not yet been performed. 8,9,11,12,15,16 In the present study, we confirmed severe vacuolar changes in the cerebral cortex and cerebellar Purkinje cell layer as well as severe astrogliosis in the cerebral white matter. Ultrastructurally, these vacuoles contained electron-dense granules and mitochondria, and some vacuoles were contiguous with the cytoplasm of degenerated cells. Furthermore, we detected a missense mutation (c.859G>C) in exon 6 of the ASPA gene and an increased concentration of NAA in urine (case 2). Therefore, we proposed a diagnosis of feline spongy encephalopathy associated with a mutation in the ASPA gene for the present cases, analogous to human CD.
Clinically, 3 distinct groups of human CD have been identified: the congenital form with severe symptoms in the first few weeks of life; the infantile form (most common), in which the disease is apparent by 6 months of age; and the juvenile form, in which the disease is apparent by the age of 4 or 5 years of age. 1 Early signs of CD include irritability and hypotonia with poor head control. Common symptoms of CD include head lag, macrocephaly, hypotonia, dysstasia, inadequate visual tracking, poor sucking ability, and intellectual disabilities. Cortical blindness and optic nerve atrophy accompany seizures in the later stages. 13,19 In the present study, affected cats showed gait disturbance and head tremors in the early stage, followed by dysstasia, intension tremors, and seizures. Therefore, the clinical course of the affected cats was similar to that of the infantile or juvenile forms of human CD. Macrocephaly, one of the diagnostic features of CD in humans, was not detected in cats. Although all affected cats had the same mutation in the ASPA gene, the clinical onset varied between 1 and 19 months of age, and they died between 7 and 53 months of age. These results suggest that intrinsic and extrinsic factors affect the onset age and survival time of each individual.
Genetic analysis identified the homozygous point mutation (c.859G>C) in exon 6 of the ASPA gene in affected cats. This mutation results in an amino acid substitution of a conserved alanine to proline (A287P). Furthermore, in silico analysis using SIFT revealed that the mutation was not tolerant. In humans, more than 70 mutations have been identified in the ASPA gene (http://www.hgmd.org), and most of them are single base-pair changes in the coding region that generally result in the loss of ASPA enzymatic activity because of the disruption of a critical functional domain in the protein. 22 Although the c.859G>C (A287P) mutation has not yet been reported in humans, a c.859G>A (A287T) mutation was identified as the cause of CD in German patients. 26 Two types of mutations, c.854A>C (E285A) and c.693C>A (Y231X), account for 97% of chromosomal mutations among Jewish patients. 17 The c.914C>A (A305E) missense mutation is the most prevalent among English, Dutch, and German patients, and affects 50% of patients with Western European ancestry. 10,14 We speculate that the c.859G>C (A287P) mutation is prevalent among domestic cats in Japan, and the identification of this mutation has enabled us to estimate the carrier frequency in the feline population.
Grossly, human CD is characterized by ill-defined demarcation between white and gray matter of all lobes in the cerebrum. 1 In the present study, the affected cats exhibited a lack of distinction between deep cortex and subcortical white matter of affected lobes in the cerebrum. The gross findings are comparable among humans and cats. On the other hand, histopathological findings in the present study slightly differed from those of typical human CD. Human CD is histopathologically characterized by extensive intramyelinic vacuole formation in the white matter of the brain as a result of the splitting of myelin lamellae at the intraperiod lines, and moderate to severe myelin paucity; myelin splitting may occur in all parts of the myelin sheath. Furthermore, the deep cortex contains degenerated astrocytes with swollen cytoplasm containing enormously elongated mitochondria with abnormal cristae. 1 In contrast, the affected cats did not show significant vacuole formation in the cerebral white matter, while the vacuoles were predominantly distributed in the cerebral cortex and Purkinje cell layer of the cerebellum. Gemistocytic astrocytes were present in the cerebral white matter. Ultrastructurally, the cerebral cortex contained numerous intracytoplasmic vacuoles containing mitochondria that were not elongated, and a few intramyelinic vacuoles. Therefore, the majority of vacuoles were not associated with myelin sheaths. Immunohistochemistry revealed that the wall of vacuoles and contiguous neuropil were positive for neurofilament and some cells in or around the vacuoles were positive for MBP and Olig2. The results suggest that vacuolar changes were caused by intracytoplasmic edema in neurons and oligodendrocytes. In the mouse model of CD that carries null alleles of the ASPA gene, the distribution of vacuoles varied between different strains. Vacuolization was predominant in the subcortical white matter of the cerebrum and Purkinje cell layer of the cerebellum in ASPATm1Mata mice. 18 In ASPAnur7 mice, vacuolization was most severe in the brain stem and cerebellar white matter. The cerebral cortex and subcortical white matter were also affected. 7,23 In ASPAlacZ/lacZ and ASPAdeaf14 mice, vacuolization was most severe in the cerebral cortex and brain stem as well as in the cerebellar Purkinje cell layer. 6,20 Therefore, in addition to species differences, the histopathological difference between humans and cats may be due to the expression levels or activities of ASPA associated with mutations in the ASPA gene. NAA, which is synthesized exclusively in neurons, is present at high concentrations, and a very high NAA gradient is normally maintained between neurons and the surrounding extracellular fluids. 4 NAA is constantly being released, with its obligated water, to extracellular fluids. 5 One possible mechanism suggested in human CD is the accumulation of the osmolyte NAA in brain extracellular fluids due to ASPA deficiency inducing the deconstruction and splitting of oligodendrocyte myelin sheaths at their extracellular fluid intraperiod lines, with the formation of large fluid-filled vacuoles around neurons and astrocytes. 2,3 However, the present results suggest that cats carrying the c.859G>C mutation in the ASPA gene had vacuolar changes due to intracytoplasmic edema in neurons and oligodendrocytes. Therefore, an increase in cytosolic components may be important for vacuole formation in feline spongy encephalopathy associated with ASPA deficiency. Various hypotheses regarding the metabolism of NAA and its possible role in the central nervous system have been proposed, but the pathophysiology of human CD still remains unclear. In the feline cases, though we speculate that NAA accumulation results in an osmotic gradient that produces intracytoplasmic edema, more detailed studies are required to clarify this hypothesis.
In conclusion, based on clinical, genetic, and pathological findings, the neurodegenerative disease in 4 mixed-breed cats was similar to the infantile or juvenile forms of human CD, and this is the first study to report feline spongy encephalopathy with a mutation in the ASPA gene. Genetic analysis revealed that all affected cats had the same missense mutation, c.859G>C (A287P), suggesting that this mutation is prevalent in the cat population of Japan and its detection may be useful for the diagnosis of young cats showing diffuse degeneration of the cerebral cortex. Differences in histopathological features between humans and cats may provide insights into other mechanisms of CD and may be useful for investigating the pathogenesis of and possible treatments for this disease.
Footnotes
Acknowledgement
We thank Dr I. Matsumoto for his excellent technical assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by a grant-in-aid from Japanese Society for the Promotion of Science (18H02338, 19J22779).