
Abstract
There is a clear link between defects in autophagy and the development of autoimmune and chronic inflammatory diseases, raising interest in better understanding the roles of autophagy within the immune system. In addition, autophagy has been implicated in the immune response to infection by pathogenic microbes. As such, there are efforts currently underway to develop modulators of autophagy as a therapeutic strategy for the treatment of the autoimmune, inflammatory, and infectious diseases. In this review, we discuss the numerous roles for autophagy in immunity and how these activities are linked to disease. We highlight how autophagy affects pathogen clearance, phagocytosis, pattern recognition receptor signaling, inflammation, antigen presentation, cell death, and immune cell development and maintenance. With these diverse and extensive immune-related functions for autophagy in mind, we finish by considering the possible implications of targeting autophagy as a therapeutic strategy.
Autophagy is a dynamic process that targets cellular cytoplasmic contents for lysosomal degradation. 54 Autophagy maintains cellular homeostasis by sequestering and degrading damaged proteins and organelles to prevent their accumulation and subsequent toxicity to the cell. Autophagy can also be induced by stimuli such as prolonged starvation, where it degrades proteins to replenish amino acid pools. Initiation of autophagy involves the sequestration of a small portion of the cytoplasm in a membranous sac referred to as a phagophore 39,54 (Fig. 1, panel 1). Phagophore formation is mediated by translocation of the ULK1 complex (ULK1/ULK2, ATG13, FIP200, ATG101) from the cytoplasm to the endoplasmic reticulum (ER). The ULK1 complex recruits the PI3 kinase (PI3K) complex consisting of ATG14L, BECLIN1, VPS15, and VPS34. 39,54 The PI3K complex produces phosphatidylinositol 3-phosphate (PI3P), which is not normally found in the ER and is essential for autophagy. 38,42,54 Elongation of the phagophore to generate an autophagosome depends on 2 ubiquitin-like conjugation systems (Fig. 1, panel 2). In the first system, ATG12 (ubiquitin-like protein) is synthesized with an exposed C-terminal glycine, activated by ATG7 (E1-like), and transferred to ATG10 (E2-like), which facilitates its final ligation to ATG5. ATG5 noncovalently interacts with ATG16L1, resulting in an ATG5-ATG12–ATG16L1 complex that is present on the phagophore and elongating membrane. 38,42,53,54,86 The second ubiquitin-like component is LC3 (microtubule-associated protein 1 light chain 3). 38,42,54 Upon synthesis, pro-LC3 is processed by the cysteine-protease ATG4 to generate a cytoplasmic form called LC3-I. LC3-I is conjugated to phosphatidylethanolamine, generating the membrane-bound form called LC3-II, through the actions of ATG7 (E1-like) and ATG3 (E2-like). ATG5-ATG12 facilitates LC3 lipidation through its interactions with ATG3, while ATG16L1 specifies the localization of LC3 conjugation to the autophagosome membrane. Upon completion of the autophagosome membrane, removal of external LC3 by ATG4 facilitates autophagosome maturation as well as recycling of LC3, while LC3 on the interior of the autophagosome is inaccessible to ATG4 and will be degraded. Autophagosome maturation occurs through SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor)-mediated fusion of autophagosomes with lysosomes to degrade the cargo 54 (Fig. 1, panel 3).
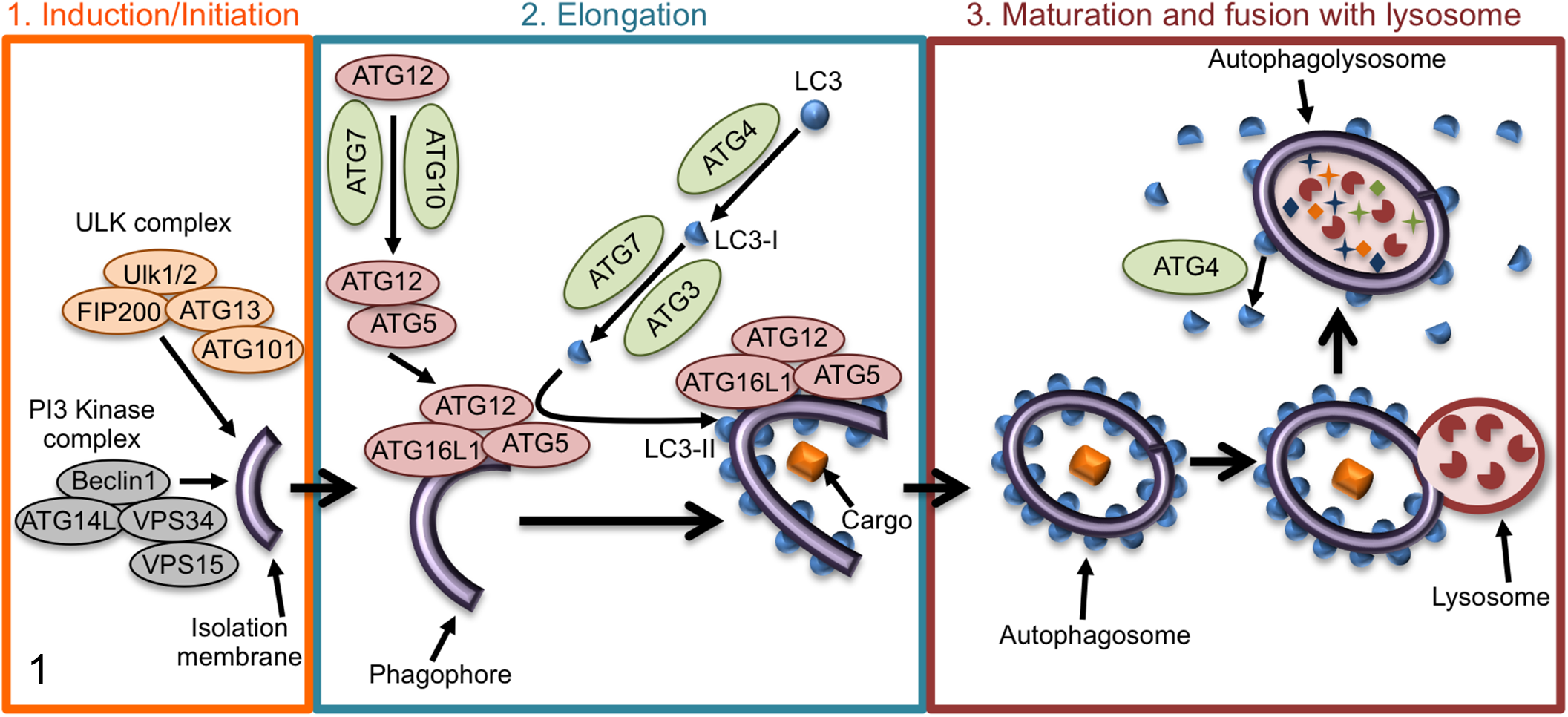
Schematic of the stages of autophagy. Panel 1: Autophagy initiates with the formation of a phagophore, which is mediated by the ULK1 and PI3 kinase (PI3K) complexes. Panel 2: Elongation of the autophagosomal double membrane depends on 2 ubiquitin-like conjugation events. First, ATG12 (ubiquitin-like protein) is ligated to ATG5. The second ubiquitin-like component is LC3, which is conjugated to phosphatidylethanolamine, generating the membrane-bound form called LC3-II. Panel 3: Upon completion of the autophagosome membrane, external LC3 is removed by ATG4 to facilitate autophagosome maturation and recycling of LC3. Autophagosome maturation occurs through SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor)-mediated fusion of autophagosomes with lysosomes to degrade the cargo. More details for each step are described in the main text.
Defects in autophagy are associated with susceptibility to autoimmune and inflammatory diseases. Specifically, polymorphisms in autophagy genes have been linked to increased susceptibility to Crohn disease, 11,22 a type of chronic inflammatory bowel disease, and the autoimmune disorder systemic lupus erythematosus (SLE). 23,26 Autophagy has also been linked to the autoimmune disorders rheumatoid arthritis and multiple sclerosis. 91 For Crohn disease, an ATG16L1 variant, T300A, is associated with a defective innate immune response to enteric bacteria, resulting in increased numbers of intracellular bacteria, 16,84 elevated secretion of the proinflammatory cytokines, 40 and impaired antigen uptake and processing. 16,81 In addition, ATG16L1 is critical for the function of Paneth cells in the small intestine, and decreased ATG16L1 expression in these cells results in defective secretion of cytoplasmic granules of antimicrobial factors and increased expression of proinflammatory signals. 12 For SLE, single-nucleotide polymorphisms in ATG5 have been associated with disease development. 23,26 Autophagy is involved in a number of processes that, when defective, could explain the susceptibility of these patients to SLE, including dead cell removal, scavenging of intracellular nucleic acid, regulation of type I interferon (IFN) responses, and control of the long-term survival of B and T cells. The roles for autophagy in these processes will be described in more detail below.
Autophagy has also been implicated in the immune response to infection by pathogenic microbes. Most studies of autophagy during infection have focused on xenophagy, a type of selective macroautophagy that specifically targets intracellular pathogens to lysosomes, restricting their replication and survival (Fig. 2). However, for all intracellular bacterial pathogens that have been investigated, there is evidence of strategies that allow them to subvert or exploit xenophagy to promote pathogenesis. 34 Infection of genetically engineered mice with mutations in the autophagy pathway has revealed that autophagy-associated proteins often function outside of xenophagy to influence immune responses to bacterial pathogens. 18,42,77 A caveat inherent to this area of research is that most studies that implicate autophagy in immunity have only looked at one gene or a handful of genes, but many of the autophagy proteins have other functions independent of autophagy. 18,34,42,77 Therefore, in many cases, whether the susceptibility of these mice is due to loss of autophagy, loss of an autophagy-independent role for the particular ATG protein, or effects on a combination of these processes remains unknown since conclusions about the role of autophagy in immunity cannot be made based on data from a single autophagy protein. With this caveat in mind, we will discuss the immune responses that autophagy and autophagy-associated proteins have been implicated in beyond xenophagy (summarized in Fig. 3) and how these diverse roles may affect strategies aimed at modifying autophagy to treat disease.
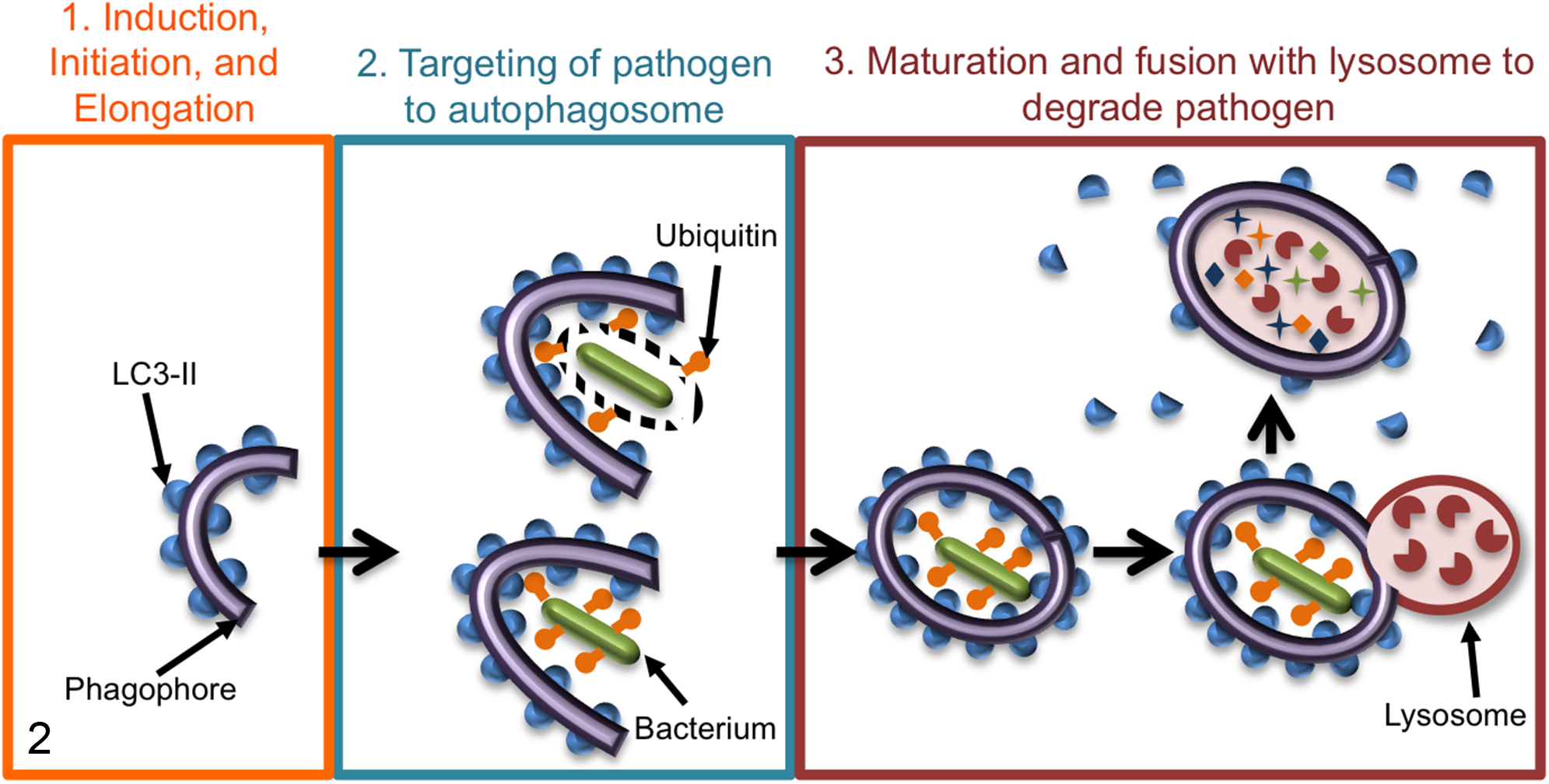
Schematic of xenophagy targeting bacterial pathogens to the lysosome for degradation. In general, once a pathogen either escapes into the cytosol or is exposed to the cytosol through damage to the vesicle in which it resides, xenophagy is induced by cytoplasmic recognition of pathogen or damage-associated molecular patterns (PAMPs, DAMPs). Panel 1: Xenophagy is initiated with phagophore formation and elongation of the autophagosomal double membrane. LC3-II (blue half-circles) is formed and localizes to the inner and outer autophagosome membranes. Panel 2: Bacteria (green rods) within a damaged vesicle (top) or within the cytosol (bottom) colocalize with ubiquitin (orange shapes), which is then recognized by autophagy receptors that also interact with LC3-II, thus targeting the bacteria to the autophagosomes. Panel 3: Autophagosomes mature and fuse with lysosomes (red circle with red partial circle shapes inside), and the cargo, including the bacteria, is degraded.
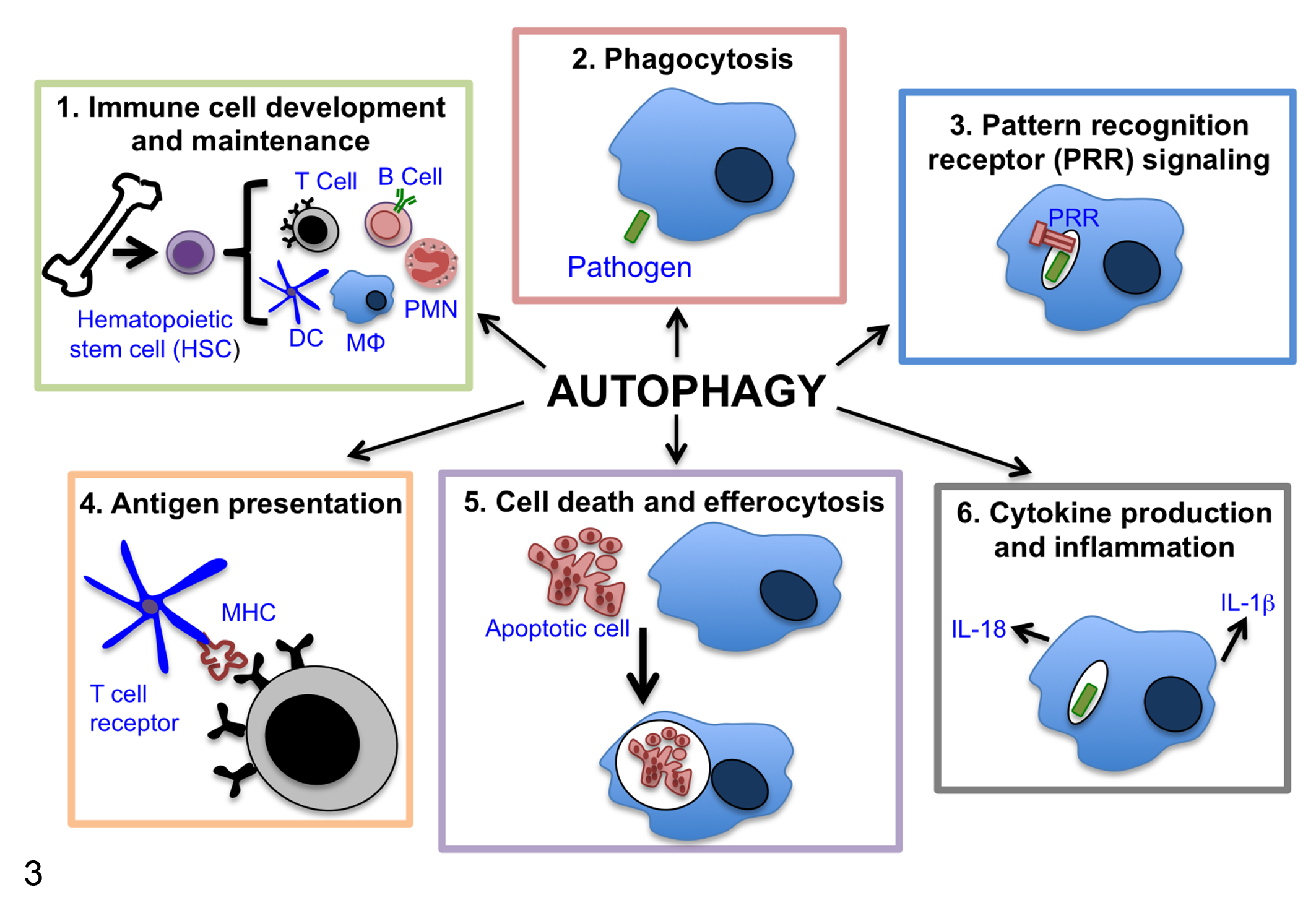
Summary of the multiple ways that autophagy proteins affect immunity beyond their role in xenophagy. Autophagy proteins are associated with and may influence immune responses by several mechanisms. Specifically, autophagy has roles in (1) immune cell development and maintenance, (2) phagocytosis, (3) pattern recognition receptor signaling, (4) antigen presentation, (5) cell death and efferocytosis, and (6) cytokine production and inflammation. These areas are discussed in detail in the main text. DC, dendritic cell; MΦ, macrophage; PMN, neutrophil.
Immune Cell Development and Maintenance
At the earliest stage of immune cell development, hematopoietic stem cells (HSCs) require autophagic clearance of damaged mitochondria (mitophagy) to maintain sufficiently low levels of reactive oxygen species (ROS) and remain in a self-renewing state. 21,45,55,56 Maintenance of HSCs is required for replenishment and function of lymphoid and myeloid progenitor cells. Accordingly, conditional deletion of the autophagy gene Atg7 or FIP200 in HSCs results in impaired self-renewal, reduced development of HSCs into common lymphoid and myeloid progenitor cells, and impaired reconstitution of hematopoietic cells in lethally irradiated mice. 45,56
Autophagy proteins also play important roles during lymphocyte development, maintenance, and activity. Using mice conditionally deleted for Atg5 at different stages of B-cell development, it was shown that Atg5 is dispensable for pro- to pre-B-cell transition but necessary to maintain normal numbers of peripheral B cells. 3 In addition, B cells deficient in Atg5 exhibit an impaired plasma cell response and reduced antibody generation in both short and long-lived plasma cells. 3,15,63 Autophagy deficiency in thymocytes results in a reduced number of CD4+ and CD8+ T cells. 4,65,80 In mature, naive T cells, autophagy deficiency results in dysregulation of cell homeostasis and enhanced cell death, which affects CD8+ T cells more than CD4+ T cells, possibly due to the higher basal level of autophagy in CD8+ T cells. 30,61,65,80,89 Loss of autophagy in mature T cells also results in reduced proliferation upon T-cell receptor stimulation. 61,65,80 T-cell-specific deletion of either Atg5 or Atg7 also resulted in reduced viability of CD8+ effector T cells and a block in CD8+ memory T-cell differentiation during viral infection. 66,73,90 In addition, deletion of Atg7 or Atg5 in regulatory T cells (Tregs) leads to decreased FOXP3 expression, increased apoptosis, decreased numbers of Tregs, and systemic inflammatory disorders, 61,87 demonstrating the importance for autophagy in the homeostasis of Treg cells and subsequent immune regulation. It is likely that the mechanism by which autophagy affects the development and maintenance of lymphocytes involves the fundamental need for autophagy to recycle damaged organelles and maintain metabolic homeostasis.
Phagocytosis
One way innate immune cells facilitate host defenses is by ingesting and eliminating pathogens through the process of phagocytosis. Immune cells also phagocytose damaged cells and debris to facilitate wound healing and tissue remodeling. Autophagy proteins can both negatively and positively affect phagocytosis. When autophagy is downregulated, the adaptor protein p62 accumulates and interacts with KEAP1 to release the transcription factor Nrf2, which activates transcription of scavenger receptors that increase phagocytosis of certain bacteria. 8 Many autophagy proteins also participate in LC3-associated phagocytosis (LAP), which traffics ingested pathogens and apoptotic cells to the lysosome for degradation. In contrast to xenophagy, LAP uses RUBICON and UVRAG instead of ATG14L in the PI3K complex, does not depend on ULK1, and results in LC3+ single-membrane vesicles instead of the double-membrane characteristic of autophagosomes. 25 Because LAP was discovered relatively recently, previous studies describing pathogen-associated LC3+ vesicles now require additional investigation to determine if these are generated through xenophagy or LAP. Nonetheless, most pathogen defenses that block clearance by xenophagy will also inhibit LAP, and the specific roles for LAP in controlling intracellular pathogen replication in vivo require further investigation.
Pattern Recognition Receptor Signaling
Following infection of a host cell, autophagy and LAP can increase pattern recognition receptor (PRR) signaling by delivering pathogen-associated molecular patterns (PAMPs) to PRRs called Toll-like receptors (TLRs) within endosomes. 19,25 MAP1S, a protein that interacts with autophagosome-associated LC3 to regulate autophagosomal biogenesis and degradation, also interacts with the TLR adaptor MyD88 to further enhance TLR signaling. 76 TLR signaling results in the activation of transcription factors such as nuclear factor (NF)–κB and interferon-regulatory factors (IRFs), which leads to the induction of innate immune responses that clear infecting microbes by producing inflammatory cytokines, type I IFN, and other immune mediators. TLR signaling is also involved in activation of antigen-specific adaptive immune responses. 37,41,93 Although the role of autophagy in regulating TLR signaling in T cells is unknown, autophagy in activated B cells affects B-cell proliferation, antibody production, and antibody class switching through regulation of TLR signaling. 2 Importantly, autophagy has also been shown to be important for ultimately trafficking TLR signaling components to lysosomes for degradation where signaling is terminated, which can be critical for preventing prolonged responses and autoimmunity. 2,27,41
Autophagy can also repress RIG-I-like receptor (RLR) signaling through its role in maintaining mitochondrial integrity. RLRs are PRRs that sense PAMPs within foreign (ie, viral) RNA and induce antiviral immune responses. In the absence of autophagy, cells accumulate damaged and dysfunctional mitochondria that produce high levels of reactive oxygen species (ROS) and lead to increased RLR signaling-induced type I IFN production. 82 In addition, through autophagy-independent mechanisms, the ATG5-ATG12 conjugate directly associates with and negatively regulates RIG-I, and ATG9A negatively regulates DNA receptor STING (stimulator of IFN genes), both leading to a decrease in type I IFN responses. 31,71 The effects of changes in type I IFN signaling depend on the pathogen. For instance, type I IFN is critical for host defense to most viral infections but can be either detrimental or protective to the host during bacterial infections, depending on the pathogen. 9
Inflammation
The regulation of PRR signaling by autophagy affects downstream inflammatory pathways. Inflammatory responses are critical for controlling pathogen replication and spread but can also cause extensive tissue damage and, therefore, must be tightly regulated. Autophagy also regulates inflammation by repressing inflammasome activation. 18,42,77 The inflammasome is a protein complex that forms upon activation of PRRs and consists of nucleotide-binding oligomerization domain (NOD)-like receptors, the apoptosis-associated Speck-like protein (ASC) adaptor protein, and pro-caspase-1. 10 Pro-caspase-1 is cleaved into its active form, resulting in the release of the proinflammatory cytokines interleukin (IL)-1β and IL-18 while also inducing pyroptotic cell death. IL-1β, a potent proinflammatory molecule that is typically important for control of bacterial infections, can also lead to significant immunopathology that can ultimately be detrimental to the host. 70 Loss of autophagy in myeloid cells results in higher levels of inflammasome activation and secretion of IL-1β and IL-18, which can lead to inflammation-dependent tissue damage. 1,13,32,72 The mechanisms of autophagy-based suppression of inflammasome activation may be indirect and rely on autophagy-dependent degradation of antigen, degradation of inflammasome components, 24,75 or mitochondrial homeostasis. 57,94 Recently, it was reported that although basal autophagy inhibits IL-1β secretion, inducing autophagy promotes IL-1β secretion from macrophages through an inflammasome-, GRASP55-, and Rab8a-dependent mechanism. 20 Therefore, more studies are necessary to understand the complex roles for autophagy in regulating IL-1β secretion.
Antigen Presentation by Major Histocompatibility Complex
In addition to regulating initial antigen sensing and the resulting inflammatory responses, autophagy also affects the efficiency of major histocompatibility complex (MHC) presentation. MHC molecules present processed antigen to T cells to trigger T-cell activation, which is essential for T-cell-mediated adaptive immune responses. MHC class I (MHC-I) presents antigens derived from the cytosol to CD8+ T cells while MHC class II presents antigen from endosomes and lysosomes to CD4+ T cells. Autophagy and LAP are directly involved in MHC-II antigen presentation in antigen-presenting cells (APCs) such as macrophages, dendritic cells (DCs), and thymic epithelial cells since autophagosomes containing degraded extracellular pathogens or intracellular antigens fuse with the MHC-II loading compartments. 29,48,58 –60,69,74 Autophagy has also been shown to promote citrullination of self-peptides and MHC-II presentation of these neo-epitopes by APCs, leading to T-cell activation and subsequent autoimmunity. 28 Although not implicated in canonical MHC-I presentation, autophagy and LAP can promote cross-presentation of extracellular antigens by MHC-I. 17,44,52,67,83,85 In contrast to facilitating MHC-II presentation and MHC-I cross-presentation, a recent study showed that DC-specific Atg5 deletion in mice resulted in extended glycolipid antigen presentation via MHC-related CD1d1 molecules and enhanced activation of invariant natural killer T (NKT) cells due to a reduced rate of calthrin-dependent CD1d1 internalization, suggesting that autophagy could downregulate CD1d1 antigen presentation. 33
In addition to direct effects of autophagy on MHC presentation of antigen, autophagy has been implicated in the migration of DCs, which is important for antigen acquisition and trafficking to secondary lymphoid organs to present antigen to T cells. In human monocyte-derived DCs, Atg16L1 knockdown and chemical inhibition of autophagy resulted in a decrease in filopodia-like extensions, a ruffled phenotype, and an increase in local adhesion sites called podosomes, all suggesting enhanced adherence and impaired motility. 88 In support of these findings, ATG5 and ATG16L1 were also shown to be important for DC migration in transwell assays and in mice. 88
Cell Death and Efferocytosis
Autophagy can directly increase host cell survival by acting as a starvation response. However, autophagy can also decrease cell survival through autophagic cell death, a type of nonapoptotic cell death characterized by autophagic vacuolization of the cytoplasm that may result from uncontrolled levels of autophagy. 46 The mechanism by which a cell dies can have a major impact on immune responses since damage-associated molecular patterns (DAMPs) released from dying cells will trigger inflammation. 36 Apoptosis is often considered immunologically silent, and apoptotic cells are rapidly taken up by phagocytes through a process called efferocytosis or endothelial cells through entosis, which further compartmentalizes pathogens, 49 resolves inflammation, 35 and promotes efficient antigen presentation. 6,7 In contrast, necroptosis and pyroptosis are lytic and release DAMPs that lead to more inflammation and tissue damage. The roles for autophagic cell death in inflammation have yet to be explored in detail, but their lytic nature indicates that they are proinflammatory. Indeed, phagocytosis of cells undergoing autophagic cell death can induce inflammasome activation and IL-1β secretion. 5,64 Notably, ATG5 also affects cell death independent of autophagy when it is cleaved by calpain and translocates to the mitochondria, where it has proapoptotic effects. 92 LAP is also important for the efficient clearance of apoptotic cells by efferocytosis, 50,51 further increasing the impact of autophagy-associated proteins on cell death and inflammation.
Summary and Therapeutic Potential
Autophagy-associated proteins have diverse effects on inflammation and immunity that depend on the pathogen and timing of the response. 34 There is growing interest in manipulating autophagy within the immune system to treat infections, autoimmunity, and inflammatory diseases. Given the impact of autophagy-associated proteins on diverse immune responses, pursuing autophagy as a therapeutic target requires the consideration of a number of factors and a more critical and comprehensive evaluation of the role of autophagy-related proteins in immunity to understand the consequences of intervening with autophagy. In particular, manipulation of the activity of autophagy proteins may affect both autophagy-dependent and independent processes, which can have a myriad of effects on disease outcomes. In addition, inducing autophagy may require increasing protein activity, as opposed to the more standard therapeutic approaches of inhibiting protein activity. Nonetheless, multiple strategies are currently in development, including the repurposing of pharmacologic agents that are approved by the Food and Drug Administration and have pleiotropic effects, including the induction of autophagy. 14,47,62,68,79 The likely complications resulting from the pleiotropic effects of these pharmacologic agents have raised interest in more specific ways to modulate autophagy with limited side effects. One such approach is lentiviral- or adenoviral-based gene therapy for tissue specific expression of autophagy genes. 43 Another approach being pursued is the use of cell-penetrating autophagy-inducing peptides. 78 These areas still require a significant amount of research to fully understand their clinical potential but may hold promise for future therapeutic strategies.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: C.L.S. is supported by an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. E.M.N. is supported by an NIH NIGMS training grant T32 07200.