
Abstract
In humans and mouse models, Foxp3+ regulatory T cells are known to control all aspects of immune responses. However, only limited information exists on these cells’ role in diseases of other animals. In this review, we cover the most important features and different types of regulatory T cells, which include those that are thymus-derived and peripherally induced, the mechanisms by which they control immune responses by targeting effector T cells and antigen-presenting cells, and most important, their role in animal health and diseases including cancer, infections, and other conditions such as hypersensitivities and autoimmunity. Although the literature regarding regulatory T cells in domestic animal species is still limited, multiple articles have recently emerged and are discussed. Moreover, we also discuss the evidence suggesting that regulatory T cells might limit the magnitude of effector responses, which can have either a positive or negative result, depending on the context of animal and human disease. In addition, the issue of plasticity is discussed because plasticity in regulatory T cells can result in the loss of their protective function in some microenvironments during disease. Lastly, the manipulation of regulatory T cells is discussed in assessing the possibility of their use as a treatment in the future.
Keywords
Prior to the mid-1990s, regulatory T cells (Tregs) were described generally as “suppressor cells” because of their known suppressive functions. 17,66 Now, multiple types of these immunoregulatory cells are known to take different roles in various pathophysiological conditions, including cancer and infection, as well as homeostasis and autoimmunity. Regulatory cells are broadly characterized into the following types: CD4+ T cells (Tregs, Tr1, and Th3), 20,55,62,86 CD8+ Tregs, double-negative CD3+ T cells, γδ T cells, natural killer T cells, regulatory B cells, and myeloid-derived suppressor cells. 69,73
The cells that are the focus of this review, CD4+ Tregs, are further characterized as thymus derived (tTregs) and peripherally induced (pTregs). As their name suggests, tTregs arise in the thymus during the normal process of T lymphocyte maturation; tTregs are self-reactive, and their main role is to prevent autoimmune disease. 84 Peripherally induced Tregs are also known as adaptive or converted Tregs because they are derived from naïve CD4+ T cells in the periphery, and when these CD4+ T cells are exposed to specific cytokines, antigens, and stimulatory conditions, they become Tregs. 9 Therefore, pTregs have a more important role than tTregs during infection, and tTregs and pTregs differ slightly with regard to cellular stability and plasticity. 48,50,84 Recently, neuropilin 1 was discovered as a marker of pTreg differentiation. 90,93
Depending on the context of disease, Tregs can be either beneficial or detrimental. Certain Tregs and their balance with effector T cells can be exploited for therapeutic purposes. 1,47,59,64,65,82,95 Most Treg research has been done in mice and humans; however, in recent years, multiple studies regarding Tregs in healthy and diseased animals have been performed. In this review, we discuss tTregs and pTregs and their presence and roles in animal disease. We focus on CD4+ Foxp3+ T cells, which currently make up the most characterized subset of all regulatory cells. 63
How Do Tregs Function?
Much of the knowledge regarding the function of Tregs comes from in vitro and in vivo mouse experiments. Tregs are generated under physiological and pathological conditions, 69 and they act by either direct regulation of T effector cells or indirect regulation by blocking the function of antigen-presenting cells (APCs), which prevents the activation of effector T cells. APCs are a group of immunocompetent cells that mediate cellular immune responses by processing and presenting antigens to T cells and inducing their activation; dendritic cells (DCs) are the main antigen-presenting cells. In most cases, the mechanism employed by Tregs depends on the disease setting, the targeted cell, the local inflammatory microenvironment, and the anatomical location. 87 All of these mechanisms are summarized in Table 1 and Fig. 1.
Treg Cell Suppressor Mechanisms.

Abbreviations: ADP, adenosine diphosphate; APCs, antigen-presenting cells; ATP, adenosine triphosphate; CD, cluster of differentiation; COX, cyclooxygenase; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; DCs, dendritic cells; DR5, death receptor 5; IDO, indoleamine 2,3- dioxygenase; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; LAG-3, lymphocyte-activation gene 3; LFA-1, lymphocyte function-associated antigen 1; MHC-II, major histocompatibility complex class II; Nrp-1, neuropilin 1; Rc, receptor; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
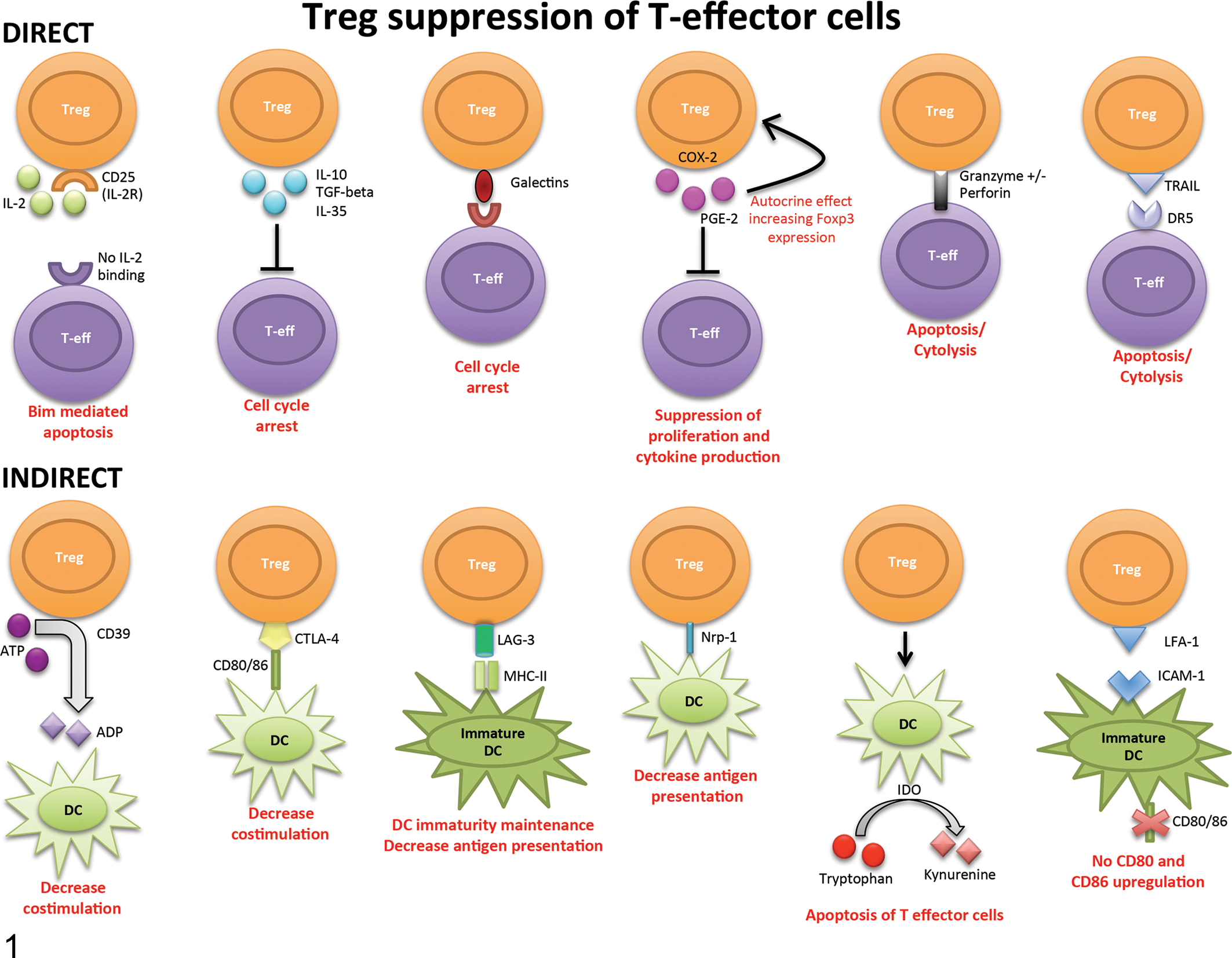
Major mechanisms by which Treg cells can directly or indirectly suppress effector T cells. Tregs may directly suppress effector T cells or indirectly suppress them by interacting with APCs/DCs. ADP, adenosine diphosphate; APCs, antigen-presenting cells; ATP, adenosine triphosphate; CD, cluster of differentiation; COX, cyclooxygenase; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; DCs, dendritic cells; DR5, death receptor 5; IDO, indoleamine 2,3- dioxygenase; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; LAG-3, lymphocyte-activation gene 3; LFA-1, lymphocyte function-associated antigen 1; MHC-II, major histocompatibility complex class II; Nrp-1, neuropilin 1; R, receptor; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
Direct regulation happens when Tregs secrete suppressor cytokines/molecules that can directly inhibit the function of effector T cells. Other mechanisms that cause direct death of the effector T cell are also considered direct mechanisms even if the Treg is not directly contacting/interacting with the targeted T effector cell. One of the most common mechanisms for direct suppression is the competition for IL-2. Treg cells express CD25 (IL-2 receptor) on their surface. IL-2 is needed for proliferation of Tregs as well as T effector cells. By consuming IL-2, T effector cells are deprived of this molecule required for T cell expansion. Lack of IL-2 causes Bim-mediated apoptosis in deprived effector T cells. Another direct mechanism that targets effector T cells is the secretion of anti-inflammatory molecules such as IL-10, TGF-beta, and IL-35 by Tregs. These molecules have been shown to cause cell cycle arrest. IL-10 is the most important anti-inflammatory molecule of the 3, especially in in vivo studies. 49 Other known direct mechanisms that Tregs use to control effector T cells include the expression of galectins, prostaglandin E-2, and granzymes. Cell cycle arrest results from expression of galectins, which are a family of endogenous carbohydrate binding proteins that have recently emerged as important players in the regulation of immune responses and inflammation. These proteins bind to multiple receptors. Specifically, galectin 1 on Tregs can bind to CD45, CD43, and CD7 on effector T cells and cause cell cycle arrest. Moreover, mouse studies have shown that galectin-1–deficient mice have reduced Treg activity. 72 Tregs also express cyclooxygenase-2, which induces the secretion of prostaglandin E-2. 35 It has been demonstrated that prostaglandin E-2 secreted by Tregs can act in a paracrine manner to inhibit effector T cell responses and at the same time act in an autocrine manner inducing the expression of Foxp3. In another direct mechanism, Tregs express granzymes and cause granzyme-mediated apoptosis/cytolysis of effector T cells. The tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expressed on Tregs also mediates the apoptosis/cytolysis of T effectors by interacting with death receptor 5, a member of the tumor necrosis factor receptor subfamily expressed on effector T cells. 58 Because these anti-inflammatory molecules and galectins need to bind to receptors on the targeted effector cells, Tregs need to act in a close proximity to the targeted cell.
In addition to direct suppression mechanisms, effector T cells are indirectly regulated by suppression of APCs (indirect suppression). By suppressing APCs, Tregs indirectly stop Foxp3− effector T cell activation. 72 As with direct regulation, there are multiple indirect mechanisms. Tregs can express on their surface CD39, a molecule that inactivates extracellular adenosine triphosphate (ATP) by catabolizing it into adenosine diphosphate. Because ATP is among those molecules that are released by cell damage, it can serve as a damage-associated molecular pattern molecule (DAMP). Multiple DAMPs, including ATP, function as indicators of tissue destruction by exerting inflammatory effects on DCs and other APCs. Therefore, catalytic inactivation of extracellular ATP by CD39 prevents antigen presentation and effector T cell activation. Another indirect mechanism of action is via Treg surface expression of cytotoxic T-lymphocyte-associated protein 4, which interacts with CD80 (B7-1) and CD86 (B7-1) on APCs. Interaction of cytotoxic T-lymphocyte–associated protein 4 with CD80 and CD86 on APCs blocks the subsequent increase of CD80 and CD86 expression or even downregulates CD80 and CD86 expression. In normal conditions, CD80 and CD86 are costimulatory molecules expressed on the surface of APCs to promote T effector cell activation. Comparably, lymphocyte-activation gene 3 on Tregs can interact with the major histocompatibility complex class II on APC, maintaining APC immaturity and inhibiting antigen presentation to T effector cells. Neuropilin-1 is not only a marker of differentiation between tTregs and pTregs but is also used by Tregs to interact with APCs. Expression of neuropilin-1 by Tregs causes a longer interaction between Tregs and APCs, thereby limiting antigen presentation to T effectors. Another suppressive molecule with a major effect on APCs, and specifically on DCs, is indoleamine 2,3- dioxygenase (IDO). 39 Tregs can stimulate DCs to express IDO, and this expression catabolizes the conversion of tryptophan to kynurenine, a toxic/proapoptotic molecule for effector T cells. Lymphocyte function-associated antigen 1, an adhesion molecule expressed on Tregs, can bind to intercellular adhesion molecule 1 on immature DCs, preventing upregulation of CD80 and CD86. 14 The previously mentioned anti-inflammatory molecule IL-10 not only has a direct suppressive role on T effector cells but can also downregulate co-stimulatory molecules on DCs and suppress their synthesis of pro-inflammatory cytokines, thereby blocking APC function. 62
It is important to consider, as mentioned previously, that most of these data are from studies that have hypothesized how Tregs could function based on in vitro experiments; therefore, many of the potential mechanisms might not occur in vivo. It is also important to understand that Tregs most likely use multiple mechanisms of action simultaneously. 72,84,87 Regarding antigen specificity, even though some studies have shown that antigen-specific Tregs suppress effector T cells more effectively than nonspecific Tregs, 75 they can also function in a nonspecific manner 69 as long as they are activated.
Treg Plasticity and Stability
The term Treg plasticity implies that within the Treg subset, there are subpopulations that maintain their core Treg identity (expression of Foxp3) but that have a malleable nature that allows them to suppress all different types of T effector cells. It was recently discovered that for Tregs to be functional, they need to express not only the Foxp3 transcription factor but also transcription factors, microRNAs, and chemokine receptors of the T effector cells they are targeting (Fig. 2). For example, Tregs that specifically suppress Th1 effector cells not only express Foxp3 but also the Th1 transcription factor (Tbet) and the Th1 chemokine receptor (CXCR3). By expressing the chemokine receptors of the targeted cells, Tregs can migrate to the site of inflammation where they exert their suppressive activity and recognize those effector T cells they are meant to suppress. Similar to Tregs that suppress Th1 effector cells, expression of Th2, Th17, or T follicular helper effector cell transcription factors, chemokine receptors, and micro-RNAs occurs in those Tregs that specifically suppress Th2, Th17, or T follicular helper effector cells (Fig. 2). Stability, on the other hand, is another important characteristic to take into account. As previously mentioned, Tregs are imprinted with Foxp3. Foxp3, which encodes the transcription factor scurfin, is a master regulatory gene for the development, stability, and regulation of signaling molecules and modulation of micro-RNA and epigenetic machinery to maintain Treg identity, function, and stability. 68 Multiple studies have shown that under extreme inflammatory environments, Tregs can lose Foxp3 expression and their suppressive capacity, becoming ex-Tregs with features of T effector cells. This implies that Tregs can be reprogramed into T effector cells, shifting from an anti-inflammatory/immunosuppressive (Treg) population to a pro-inflammatory one (T effector cell). In many instances, it is believed that pTregs are less stable than tTregs. 46,52 However, in vitro and in vivo analyses have shown a propensity of Tregs to shift to mainly Th17 and T follicular helper cells, and this was independent of thymic (tTreg) or peripheral (pTreg) origin. 92,94 Some have hypothesized that in the case of Tregs, this may represent an extreme adaptability of Tregs to provide an immediate effector-type response against infectious microorganisms. However, considering that Tregs recognize self-antigens, this could have serious consequences for the host by causing severe tissue damage or contributing to autoimmune disease. While in vitro studies have shown that the presence of TGF-β and retinoic acid can help preserve Tregs, 6 these 2 molecules are unlikely to be present in inflammatory microenvironments. Therefore, it is hypothesized that after Tregs initially access inflammatory locations, they can contribute to immunopathology. 84
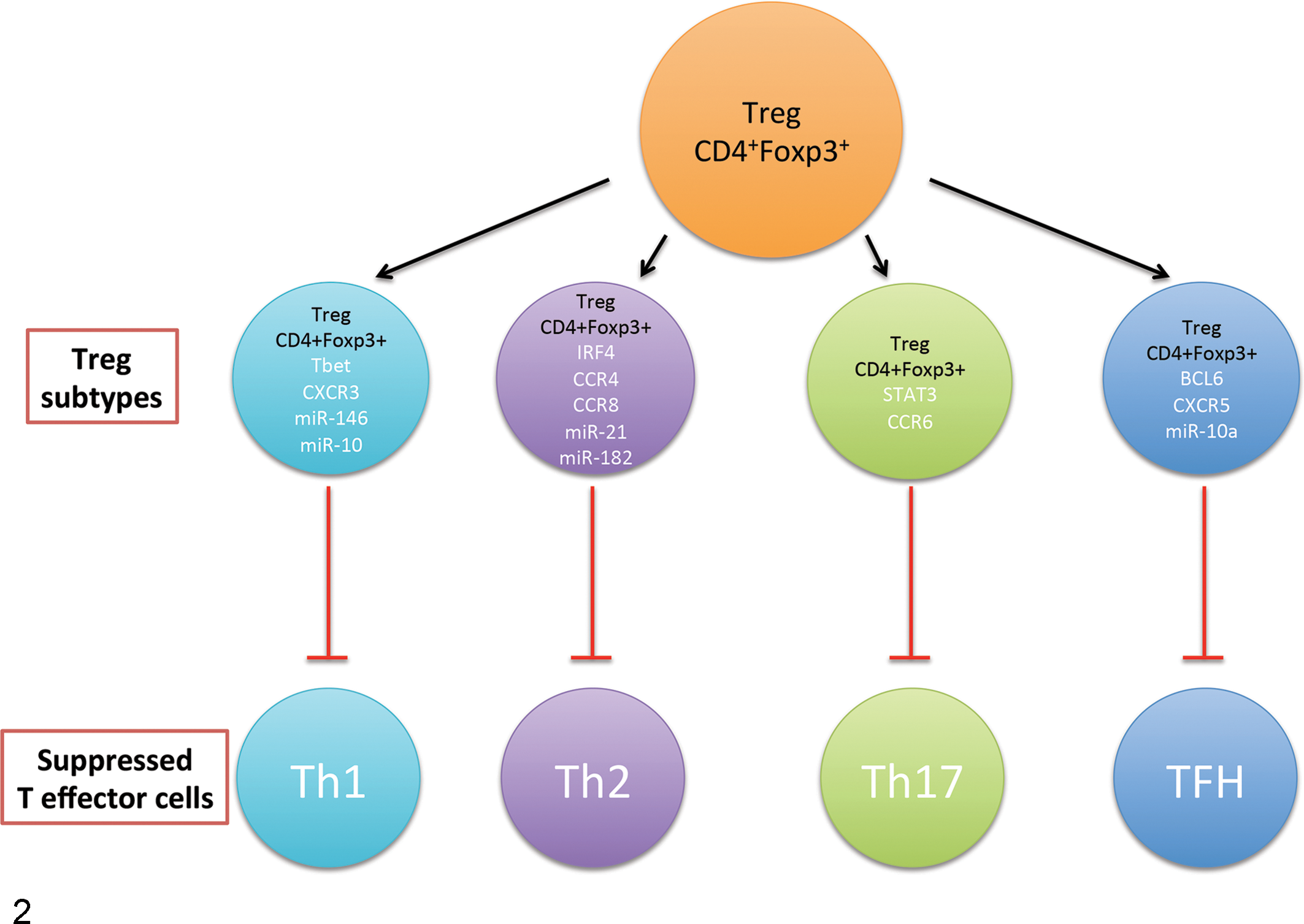
Treg plasticity. Tregs can express the same transcription factors, chemokine receptors, and microRNA-signatures as the effector cells they suppress. BCL6, B-cell lymphoma 6; CCR4, CCR6, and CCR8, CC chemokine receptor type 4, 6, and 8; CXCR3 and CXCR5, CXC chemokine receptor type 3 and 5; IRF4, interferon regulatory factor 4; miR, MicroRNA; STAT3, signal transducer and activator of transcription; TBET, T-box transcription factor TBX21. *TBET, IRF4, STAT-3, and BCL6 are transcription factors for those specific genes.
Because Tregs have an anti-inflammatory role in different circumstances (autoimmune disease, infection, and cancer), Treg stability can be either beneficial or detrimental, depending on the setting. In the inflammatory context of an infectious disease, the shift of Tregs to ex-Tregs with an effector phenotype will probably contribute to faster elimination of the infectious microorganism. On the other hand, effector/inflammatory immune responses would not be properly controlled and the chance of immunopathology would increase, especially since those ex-Tregs could recognize self-antigens. In the case of autoimmune diseases, the reduction in the amount of Tregs would increase immunopathology, with ex-Tregs contributing to tissue damage. However, in cancer, the shift of Tregs to a pro-inflammatory population would be beneficial since pro-inflammatory responses are necessary for elimination of neoplastic cells. Because of this known plasticity, potential lack of stability, and the fact that Tregs have potentially self-reactive T cell receptors, their use in therapeutics is controversial since a shift to a pro-inflammatory subset could also cause autoimmune disease.
The shift can also happen in the other known T effector subsets, such as Th1, Th2, Th17, and others. 68 Previously, it was thought that T helper cells were committed to their specific subset. CD4+ T helper cells were divided into different subsets defined by their cytokine production and functions once those cytokines were activated. In addition, the cytokine milieu in which naïve T cells were activated used to be key to deciding what T cell subset they would become. However, recent studies have disproven this hypothesis in all T helper cell lineages. 94 T helper cell phenotypes, including Tregs, are more complicated than initially thought. Most importantly, the use of Tregs for therapeutics is more complicated than previously imagined.
Regulatory Cells in Animal Disease
Most Treg research comes from human and mouse model studies. However, in recent years, numerous publications on veterinary species, especially dogs, have emerged. Still, most of the studies in non–laboratory animals are quantitative and not mechanistic. To understand where Tregs are measured, one has to understand where Tregs are generated and/or work. Because Tregs can suppress T effectors by directly interacting with them and by indirectly interacting with APCs as described previously (section “How Do Tregs Function?”), it should be evident that Tregs can act at the sites where T lymphocytes proliferate (secondary lymphoid tissue, especially lymph nodes). In lymph nodes, Tregs can inhibit immune responses by directly suppressing T effectors as well as inhibiting antigen presentation, as previously described. Tregs also accumulate at sites where immune responses are requested (site of infection, neoplastic sites, sites of inflammation), and therefore quantification of Tregs at these sites is important. 2 Evaluation of Tregs and T effector cell numbers and/or frequencies in blood are also of interest since lymphocytes use blood to reach their final destination. Moreover, the use of histology and immunohistochemistry can be helpful in recognizing Tregs in these final sites. 31
Tregs in Healthy Animals
Even before Tregs were first recognized in the 1990s, results from human and animal studies suggested the presence of a suppressor T cell population. 88,89 In dogs, a spontaneously active population of suppressor cells in peripheral blood was suggested. 13 In the early 1980s, studies in cats also reported a suppressor type of cell. 32,33 In cows, a suppressor population was induced by the exposure of lymphocytes to concanavalin-A (con-A, a cell-surface binding lectin). 67,77 However, evidence suggests that in cows, the expression of Foxp3 might not be specific for CD4+ Tregs due to the presence of large numbers of γδ T cells and the capability of γδ T cells to have a regulatory role. 16,23,71 In sheep, con-A–induced suppressor cells were also described in the mid-1980s, 10 and these used different immunoregulatory mechanisms compared to bovine con-A–induced suppressor cells. Ruminants are not the only species to have a suppressor cell population after exposure of lymphocytes to con-A. Healthy and immunosuppressed horses showed the same type of population. 53 In pigs, Tregs were initially described in the context of fetal tolerance, 15 and a few years later, these Tregs were further characterized in lymphoid tissues. 27 Other species, such as primates, guinea pigs, harbor seals, and zebrafish, have also been shown to have Treg cell populations. 14,22,29,37,45,70
Tregs and Cancer
Tregs can interfere with anti-tumor immunity by decreasing T effector cell and CD8 T cell responses. In fact, tumors evade the immune system by potentiating Treg response and survival, increasing the ratio of Tregs to T effectors, and/or secreting molecules that create a microenvironment that increases or activates Tregs. 8 Inhibition of the development of effective anti-tumor immunity by Tregs is well documented in humans and in mice. In the dog, there are various publications regarding cancer, anti-tumor immunity, and Tregs. A recent study showed that the percentage of Tregs in the blood and lymph node of dogs with cancer are increased when compared to healthy animals. 3 A subsequent publication supported these findings and concluded that out of all the different types of cancer studied, the highest increase in blood Treg percentages when compared to healthy animals was in cases of carcinoma. In addition, the highest Treg:CD8 T cell ratio was in dogs with lymphoma. 51 In these reports, dogs with all tumors studied (oral melanoma, osteosarcoma, mast cell tumor, lymphoma, and soft tissue sarcoma) showed significantly higher Treg percentages when compared to healthy animals. 3,51,81 Moreover, in oral melanomas, the percentages of Tregs were significantly increased in the tumor site when compared to peripheral blood of the same animal. The authors hypothesized that tumor cells induced either local proliferation or selective migration of Tregs to tumor-infiltrated sites. Another subsequent paper revealed that increased frequencies of peripheral blood Tregs had a positive correlation with tumor stage and a negative correlation with T effector cell (Th1) and CD8 T cell frequencies in dogs with malignant oral melanomas. 25 A study done in osteosarcomas showed that lower ratios of CD8 T to Tregs in blood were associated with shorter survival times. 4 Similarly, in mammary carcinomas, Treg cells were associated with high histological grade and lymphatic invasion. In addition, the numbers of Treg cells infiltrating mammary carcinomas markedly increased in tumors with poor prognostic factors, such as high histological grade, lymphatic invasion, and necrosis. 31 In that study, immunohistochemistry was used to demonstrate Foxp3+ Treg infiltration at the tumor site and adjacent stroma. However, in a study that compared dogs with osteosarcoma to healthy dogs, no significant difference in frequencies of Tregs was reported. 60 The authors hypothesized that discrepancies between their study and the previously described studies could be due to a small sample size, use of submandibular lymph nodes (proximity to the oral cavity could promote immune reactivity that could affect Treg percentage), and the possibility that not all Tregs were functional. The use of different antibodies to identify Tregs could also explain these differences.
Animals with metastatic tumors have also shown increased percentages of Tregs in blood. 24 In more recent papers, the mechanisms of anti-cancer drugs have been studied in relationship to Treg numbers and frequency. In fact, dogs with different types of cancer treated with cyclophosphamide, doxorubicin, and toceranib have shown decreased Treg number/frequencies in blood and at tumor sites with tumor remission or increased antitumor immunity. 5,43,44 In horses, the only published report is on equine sarcoid disease; a significantly increased expression of Foxp3 and anti-inflammatory molecules (IL-10 and IL-4) in tumor-associated tissues of affected horses indicated an upregulation of Tregs with a secondary local, Treg-induced immune suppression. 36 To the author’s knowledge, there are no other domestic species in which Tregs and cancer have been studied, other than laboratory animal species as animal models of human disease, in which findings were similar to those in the dog and horse.
Tregs and Infectious Diseases
Infectious organisms are usually cleared from any animal system by humoral and cellular immune responses. With infectious diseases, the influence of Tregs can favorably affect the outcome or can be harmful to the host. 2 In general, it is the ratio of Tregs to T effector cells that plays a primary role in the outcome of the disease. In fact, the influx of Tregs typically seen with chronic infections can benefit the host by suppressing effector T cells and thereby reducing the extent of immunopathology. 2 However, modulation of excessive immune responses by Tregs can lead to pathogen survival and long-term persistence of the microorganism. Some investigators have hypothesized that infectious microorganisms may activate or induce Tregs 19,61,69 ; however, the mechanisms still need to be elucidated. Different cytokines, such as IL-2, IL-10, and TGF-beta secreted by infected or reacting cells, can also promote the generation of Tregs. Additionally, some microorganisms are known to activate latent TGF-beta or homologues to create an anti-inflammatory microenvironment that would nurture the development of Tregs since TGF-beta is one of the primary molecules involved in the development of Tregs. 18,54,56 In humans and mice, Tregs have been shown to limit the extent of immunopathology during infectious diseases. 84 From an evolutionary perspective, it makes no sense to develop a system that limits immune responses during infection. However, in many infectious diseases, it is not the pathogen that causes the lesion but instead the immune response generated against the infection. Multiple studies have shown that mice infected with different viruses and depleted of Tregs can show increased immunopathology. For example, in hepatitis C viral infection, Tregs can suppress cytotoxic T lymphocytes (CD8+ T cells) and reduce liver damage, and in West Nile virus infection, Tregs also control CD8+ T cells, thereby avoiding central nervous system pathology. In the case of herpes viral infection of the eye, mice depleted of Tregs in later stages of the disease developed more severe stromal keratitis lesions. However, transfer of Tregs to herpes virus–infected mice or use of therapies to increase the Treg population decreased stromal keratitis severity. 84 In general, in experimental infectious studies in which Tregs were depleted, the number of inflammatory cells increased, disease severity and tissue damage were amplified, and viral titers were cleared faster. 12,79,85
In domestic animal species, there are also diseases in which Tregs have been studied in the context of infection, usually by identifying Tregs by their expression of CD25 (IL-2 R) and Foxp3, and their production of anti-inflammatory molecules such as IL-10. In dogs, studies mainly focus on Leishmania species infection. However, the conclusions of these studies, similar to the human studies, are often equivocal. Some studies have shown no difference between healthy and Leishmania-infected animals, 78 others have shown that percentages of Tregs are reduced in infected dogs, 7 and others found that Treg-secreted IL-10 was increased in animals naturally infected with Leishmania spp. 74 In another study, Treg numbers were increased in the digestive tract of infected animals, 11 but reports on visceral leishmaniasis seem to be inconclusive. In general, an increased number of CD8 T cells is characteristic of infected but asymptomatic cases. Some have hypothesized that Tregs are reduced during Leishmania infection as a way to optimize activation of T effector functions, 7 but this does not clear the pathogen. A study on canine distemper virus infection demonstrated that numbers of Tregs fluctuate throughout infection. During the acute and subacute stages of disease, there was no evidence of Tregs in neuronal tissue; however, in chronic stages of disease, Tregs were increased. 57 A probable explanation for these differences is that Tregs need time to be recruited to the neuronal tissue. Moreover, the presence of Tregs in later stages of disease could affect (as previously mentioned) maintenance of the virus and reduction of immunopathology.
In cats, the most studied disease in the context of Tregs is feline immunodeficiency virus (FIV). Not only does the virus activate Tregs, 42,83 it has also been demonstrated that FIV can replicate within Tregs. 28 Moreover, Tregs have been shown to be activated during the acute phase of the infection. 40 The importance of these data lies in the fact that the virus could induce Tregs to become a virus reservoir and therefore improve virus survival and maintenance in the host. In fact, when Tregs of FIV-infected cats were depleted in vivo, anti-FIV immune responses were improved. 41 As in cats, Foxp3+ Tregs in cattle increase in blood after infection with bovine leukemia virus. 80 Similar findings have been shown in other species. In pigs, blood Foxp3+ Tregs increased in numbers (in vivo and in vitro) after infection with porcine reproductive and respiratory syndrome virus, 91 and in sheep, Tregs have been shown to infiltrate the skin of Psoroptes ovis–infected animals. 38 Because all of these are chronic and/or latent infections, it would be interesting to study some of the more acute infections in veterinary species to determine the importance of Tregs in that context.
In general, Tregs participate in the body’s reaction to many and perhaps all infectious agents by restraining exuberant immune responses; in many chronic infections, this restraint limits tissue damage and therefore benefits the host. However, Treg responses may handicap the efficacy of protective immunity.
Tregs and Other Diseases
In veterinary species, as in humans, Tregs not only have roles in infectious diseases and cancer but also in diseases of the immune system, such as hypersensitivities and autoimmunity. In mice, deletion of the transcription factor Foxp3 causes the failure of development of tTregs, leading to a fatal lymphoproliferative syndrome with multi-organ inflammation. 26 In dogs with atopic dermatitis, numbers of peripheral blood Tregs are increased when compared to healthy animals. 30 On the other hand, dogs with inflammatory bowel disease have reduced numbers of Foxp3+ Tregs in the lamina propria when compared to healthy dogs and dogs with intestinal lymphoma. 34 In horses with insect bite hypersensitivity, Tregs were reduced in ex vivo studies. 21
Future Perspectives
Immunotherapy using Tregs is becoming more important, 76 and depending on the context of the disease, the target treatment may be to promote (for inflammatory diseases) or reduce (for cancer) Tregs. Most research regarding Tregs comes from either humans or laboratory animal models of human disease. In veterinary medicine, studies are still scant, and further investigation is required. An important question is whether our current understanding of the mechanisms that Tregs use to immunoregulate in vitro can be manipulated in vivo. Could we enhance Treg cell numbers to control immunopathology in diseases such as granulomatous meningoencephalitis but avoid systemic immunosuppression? Will the enhanced Tregs survive in vivo? Will these cells be stable? The challenge is to understand Tregs and learn how to modify their function to achieve the proper balance between protection and pathology.
Footnotes
Acknowledgements
I thank Drs Linden Craig and Shelley Newman for reviewing the manuscript and providing intellectual feedback and Misty Bailey for technical editing.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.