
Abstract
Progeroid mouse models display phenotypes in multiple organ systems that suggest premature aging and resemble features of natural aging of both mice and humans. The prospect of a significant increase in the global elderly population within the next decades has led to the emergence of “geroscience,” which aims at elucidating the molecular mechanisms involved in aging. Progeroid mouse models are frequently used in geroscience as they provide insight into the molecular mechanisms that are involved in the highly complex process of natural aging. This review provides an overview of the most commonly reported nonneoplastic macroscopic and microscopic pathologic findings in progeroid mouse models (eg, osteoporosis, osteoarthritis, degenerative joint disease, intervertebral disc degeneration, kyphosis, sarcopenia, cutaneous atrophy, wound healing, hair loss, alopecia, lymphoid atrophy, cataract, corneal endothelial dystrophy, retinal degenerative diseases, and vascular remodeling). Furthermore, several shortcomings in pathologic analysis and descriptions of these models are discussed. Progeroid mouse models are valuable models for aging, but thorough knowledge of both the mouse strain background and the progeria-related phenotype is required to guide interpretation and translation of the pathology data.
Globally, the life expectancy of human beings is increasing, and estimations based on world population demographic data predict doubling of the proportion of people older than 60 years, between 2000 and 2050. 74 Although lifespan is increasing, health span (Table 1) does not increase proportionally and aging seems to be a major risk factor for development of chronic disease. 80,140 This will lead to an increasing population of elderly people who suffer from multiple chronic diseases and will impose a major economic challenge in the near future. 25,74,80 This understanding is the driving force behind the emergence of geroscience, a field of science aimed at unraveling the pathways involved in aging to identify possible targets for intervention and disease prevention. 74,80
Definitions of Aging, Health, and Healthspan.

A large number of mouse models to study mechanisms of aging have been developed and their number is continuously increasing. Mouse models have not always been generally accepted to study the complexity of aging, but currently their value to identify new pathways involved in aging is widely recognized and much of the progress in this field can be attributed to these models. 25,30,67,80 Progeroid syndromes are in general initiated by a mutation in a single gene. 37 Progeroid syndromes (Table 2) are defined as human genetic disorders that are characterized by a shortened lifespan and premature or early development of a distinct subset of biological alterations that are normally associated with advanced age. 37,68,83,135 Even though human progeroid syndromes are relatively rare, there has been much interest in studying these diseases. Progeroid mouse models are used not only to investigate human progeroid syndromes (Table 3) such as Hutchinson-Gilford progeria, Werner syndrome, and Cockayne syndrome but also to obtain important mechanistic insights into the aging process. In addition, mouse models of progeroid syndromes have become a very attractive tool to evaluate intervention strategies for healthy aging, because of their short lifespan, their relatively simple creation by single gene deletion, and their strong phenotypic overlap with normal aging lesions. Still, some controversy remains as to whether certain progeroid models truly represent accelerated aging. 91,92 This will be exemplified later in this review when we discuss the role of calcium-phosphorous metabolism in the development of a pathologic phenotype in KLOTHO-deficient mice. Nevertheless, most studies on mouse models of progeria have demonstrated that lifespan and onset of aging are chiefly dependent on the quality of DNA repair and genome maintenance (Table 4). 67,68,94 According to the current understanding, aging ultimately results from accumulated oxidative damage to cellular macromolecules, including DNA. 68,81,94 DNA damage, if not corrected, can result in irreversible mutations or loss of genetic information and is therefore considered to be more deleterious than damage in other macromolecules such as lipids and proteins. 68 Defects in the DNA repair machinery, which have been identified in progeroid syndromes, therefore accelerate onset of aging signs.
Definitions of Progeria, Progeroid, and Progeroid Syndrome.

Examples of 2 Major Human Progeroid Syndromes.

Target Genes and Molecular Mechanisms in Mouse Models of Progeria.
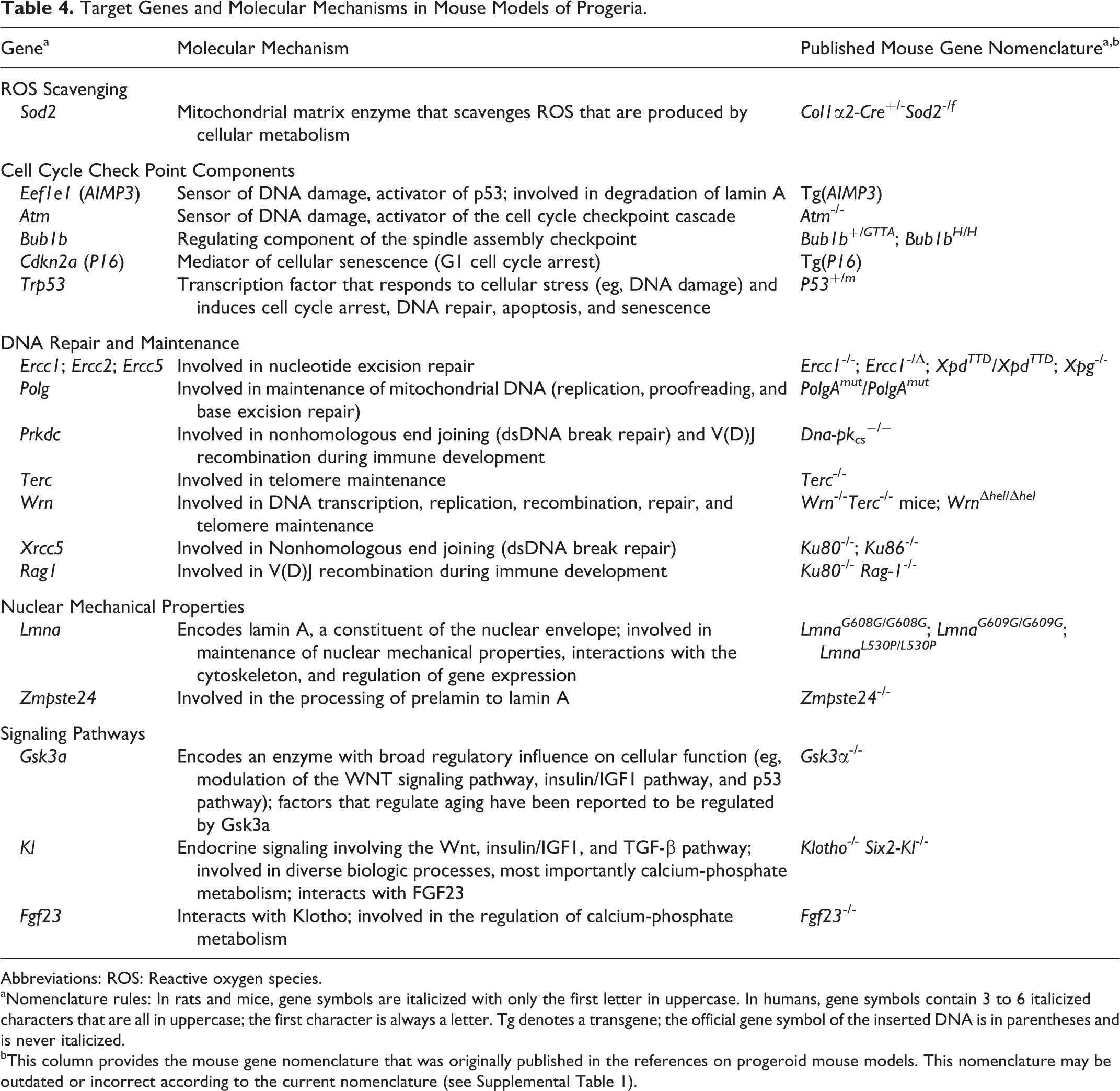
Abbreviations: ROS: Reactive oxygen species.
aNomenclature rules: In rats and mice, gene symbols are italicized with only the first letter in uppercase. In humans, gene symbols contain 3 to 6 italicized characters that are all in uppercase; the first character is always a letter. Tg denotes a transgene; the official gene symbol of the inserted DNA is in parentheses and is never italicized.
bThis column provides the mouse gene nomenclature that was originally published in the references on progeroid mouse models. This nomenclature may be outdated or incorrect according to the current nomenclature (see Supplemental Table 1).
In this review, we provide an overview of nonneoplastic macroscopic and microscopic pathology encountered in progeroid mouse models. Detailed enumeration of all the different mouse models that are available is beyond the scope of this review due to the large and ever increasing number of mouse models and often incomplete pathology data. Instead, we provide a concise reference on the phenotypes that have been most frequently reported in progeroid mouse models and that may be relevant to geroscience because of shared features with common age-related changes and syndromes in humans.
The most commonly reported phenotypes in progeroid mouse models involve bone, joint, skeletal muscle, skin, adipose tissue, cardiovascular system, nervous system, liver, kidney, and hematopoietic system. Less frequently, lesions are reported in gonads, eye, and sporadically, the gastrointestinal tract. Although a large amount of data is available on progeroid mouse models, many research papers focus on genetics and protein expression, and full pathologic examination is rarely reported. In the sections below, we have outlined in each organ system the observations made in different mouse models of accelerated aging and compared them to aging phenotypes described in geriatric mice and humans. The literature that has been reviewed and the citations often contain outdated gene nomenclature. The current nomenclature of the described progeroid mouse models can be found in Supplemental Table 1 (Table S1), and rules for standardized nomenclature can be found on the JAX nomenclature website: http://www.informatics.jax.org/mgihome/nomen/.
Bone and Joint
Common age-related lesions of the bones and joints include osteoporosis, intervertebral disc disease, degenerative joint disease (DJD) (osteoarthrosis), and kyphosis (Table 5).
Pathologic Phenotypes in Bone, Joints, Skeletal Muscle, and Nervous System of Progeroid Mouse Models.

Abbreviations: DJD, degenerative joint disease; IVDD, intervertebral disc degeneration.
aNomenclature rules: see footnote of Table 4.
Osteoporosis is a well-known bone aging phenomenon in both humans and rodents and is characterized by a reduction in bone mass and density (osteopenia) through microarchitectural deterioration. Osteoporosis increases the risk of bone fractures and is a major contributing factor to the large number of hip fractures that occur in elderly humans. 23 The major primary types of human osteoporosis are postmenopausal osteoporosis and senile osteoporosis. 84 Whereas the mechanisms that lead to the development of postmenopausal osteoporosis are relatively well characterized, the mechanisms involved in the development of senile osteoporosis are less well understood due to lack of appropriate models of physiologic bone aging. 23 As osteoporosis has also been observed in some human progeroid syndromes (eg, Werner syndrome), the genetic defects involved in these progeroid syndromes provide a starting point for research. 67 Progeroid mouse models with similar genetic defects may serve as models for senile osteoporosis. 23 Current data from progeroid mouse models suggest that senile osteoporosis results from decreased bone turnover and loss of bone volume, due to defects in osteoblastic progenitor cells, osteoblastic differentiation, or osteoblastic function. As altered bone turnover is believed to play a major role in the development of senile osteoporosis, a multimodal approach is needed to gain better insight into the phenotype. Bone matrix organization is heterogeneous and complex and can be studied at different levels (eg, organic or mineral nature, woven or lamellar texture, osteon or trabecular structure, microarchitecture and macroarchitecture). These levels cannot be examined with a single method and require different techniques. 31
A variety of methods have been used to analyze the pathophysiology, presence, and extent of osteoporosis. These methods include microcomputed tomography, densitometry, radiography, X-ray energy absorptiometry, bone marrow stem cell culture, and histologic or histomorphometric analysis. 31 Histomorphometric and functional analysis are usually not a part of routine histopathologic examination. Including these methods in future studies could provide essential information to improve the understanding of senile osteoporosis, as is demonstrated by Brennan and colleagues. 23 Dysfunction of osteoblasts, typical of senile osteoporosis, was demonstrated in progeroid Wrn -/- Terc -/- mice by histomorphometric assessment of parameters of bone formation such as osteoid deposition, mineralization surface, and mineral apposition rate. 23 Furthermore, in vitro analysis suggests that defects in osteoblastic differentiation precede osteoblastic dysfunction and reduction in bone density. 117 Dysfunction of osteoblasts is believed to play a major role in bone aging. 23
In mice, spontaneous age-associated osteoporosis is reported in several mouse strains including C57BL/6 mice and BALB/c mice. 23,157 Macroscopically, osteoporosis in animals is characterized by reduced amounts and porosity of the cancellous bone and in advanced stages leads to thinning of the cortices. These macroscopic features are difficult to appreciate in the small bones of mice. Usually in mice, reported macroscopic age-associated bone lesions are limited to kyphosis, which has been observed in normally aged C57BL/6 J mice at 2 years of age. 148 Histologic lesions with osteoporosis include reduction in number and thinning of trabecular bone and in severe cases are associated with thinning of the compact bone. Microfractures of thin trabeculae may be present. Differences in the numbers of osteoblasts and osteoclasts are dependent on the underlying cause and mechanism of osteoporosis. 142
Several progeroid mouse models develop osteoporosis early in life (Table 5). Age of onset of osteoporosis greatly varies among mouse models and supposedly depends on the underlying mechanism involved and if this mechanism is partially or completely disrupted. This is demonstrated in Ercc1 -/- mice that develop osteoporotic lesions as early as 3 weeks of age, whereas hypomorphic Ercc1 -/Δ mice show similar lesions as early as 8 weeks of age, progressing to a severe phenotype at 22 weeks of age. 33
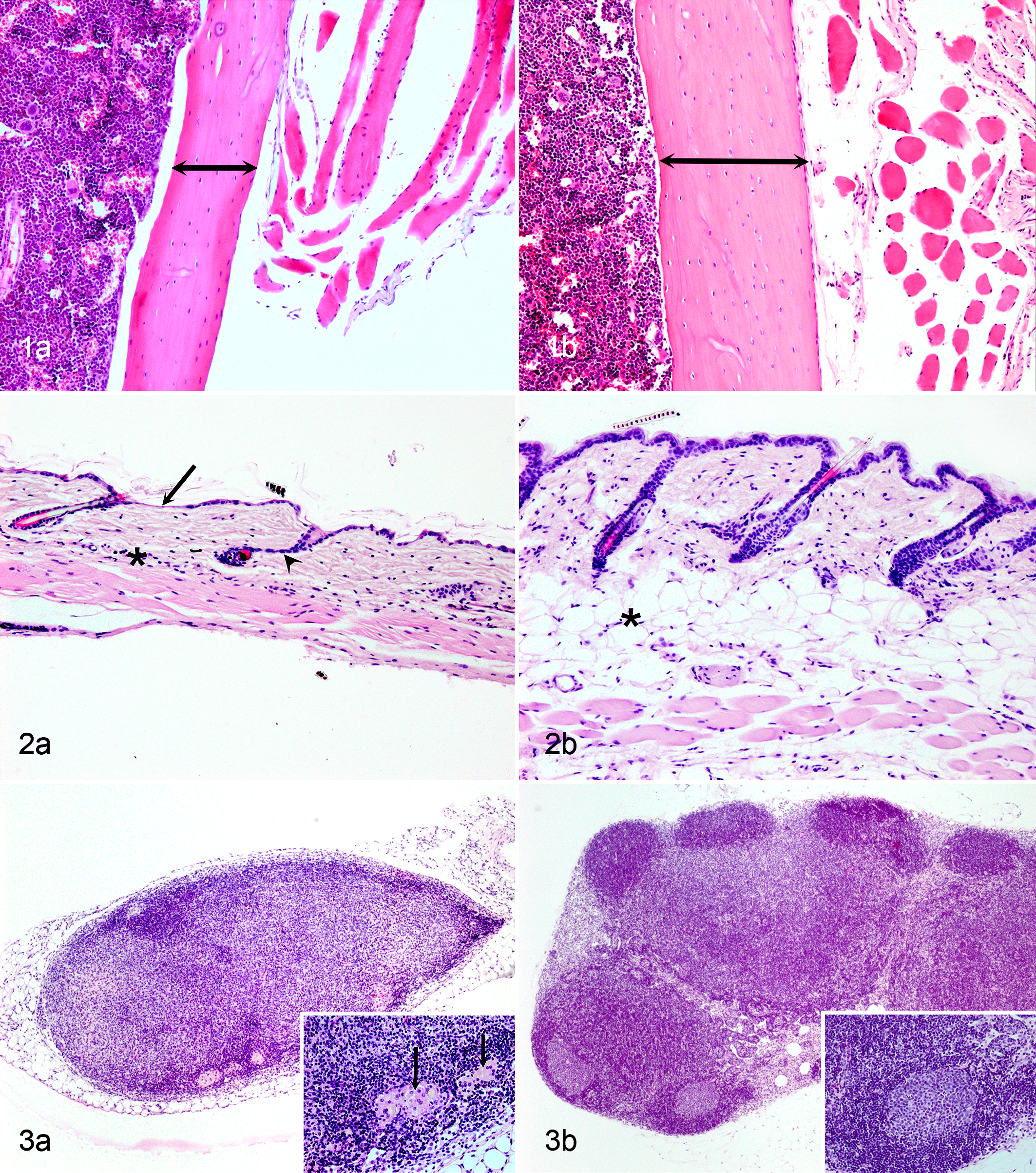
A mouse model closely mimicking the morphologic characteristics of human senile osteoporosis is the Wrn -/- Terc -/- mouse. 23 In these mice, osteoporosis is characterized by bone porosity with thinning of cortical bone, reduced trabecular bone volume with fewer and thinner trabeculae, and increased intertrabecular space, typically with a reduced number and function of osteoblasts and unchanged osteoclasts. 23 In Ercc1 -/- and Ercc1 -/Δ mice, osteoporosis was associated with atrophy of osteoblastic progenitor cells and increased numbers of senescent cells within the bone marrow stem cell population. 33 In addition, immunohistochemical expression of P16 and γ-H2AX foci was increased in osteoblasts lining the bone surface, indicating increased senescence and DNA damage, respectively. This was associated with decreased expression of the proliferation marker Ki-67 and increased levels of inflammatory cytokines interleukin 6, tumor necrosis factor α, and RANKL, which is indicative of the senescence-associated secretory phenotype. 33 Recently, a moderate reduction in the femur trabecular bone thickness (osteopenia) has been demonstrated in other progeroid mouse models in which DNA repair is perturbed, namely, in Ku80 −/− and Dna-pkcs −/− mice (Fig. 1). 123 Osteoporosis characterized by loss of trabecular bone volume, cortical thinning, and thinning of the subchondral bone plate has been reported in XpdTTD /XpdTTD mice. 19,109
Zmpste24 -/- mice, a model for Hutchinson-Gilford progeria syndrome, have also been reported to display an osteopenic phenotype. 14,124 Macroscopically, these mice had splayed, thin, and long lower incisors, fractures of the posterior portion of the zygomatic arch at 8 weeks of age, and spontaneous rib fractures near the costovertebral junction by 24 to 30 weeks of age. 14 Histologically, the spontaneous fractures were characterized by exuberant fibrous callus tissue that was markedly acellular and bone fragments lacked viable osteocytes. 14 The presence of multiple spontaneous fractures was associated with reduced bone density of ribs and vertebrae on microcomputed tomography. 14 Rivas et al 124 have demonstrated that bone marrow mesenchymal stem cells from Zmpste24 -/- mice have lost their capacity to differentiate into osteoblasts with a shift toward adipocyte differentiation. This induces a reduction in bone turnover. Histologic and histochemical analysis revealed a significant decline in bone density and architecture, characterized by a decrease in bone volume to tissue volume ratio, reduced thickness of trabeculae, decreased numbers of trabeculae, reduced numbers of alkaline phosphatase (ALP)-positive osteoblasts, reduced numbers of Toluidine blue-positive trabecular osteocytes, and also reduced numbers of tartrate-resistant acid phosphatase (TRAP)-positive osteoclasts. 124 Furthermore, there was a significant increase in the amount of bone marrow fat. 124 Another progeroid mouse model that partially mimics human senile osteoporosis is the Klotho -/- mouse. 79,157 Compared to wild type mice, Klotho-/- mice displayed reduced numbers of osteoblasts and osteoclasts per bone surface area, indicating lower bone turnover and a decrease in bone formation rate. 79 The reduced numbers of osteoclasts in the Zmpste24 -/- mice and Klotho -/- mice are in contrast to normal numbers of osteoclasts reported in multiple studies on human senile osteoporosis. 23 This illustrates that phenotypic differences in seemingly similar disease processes in mouse and human can suggest different underlying mechanisms.
Degenerative joint disease is another major debilitating syndrome that affects the elderly population and is also common in aging animals. Worldwide estimates of the affected proportion of people older than 60 years vary between 10% and 50%. 107,144 Aging changes in the articular cartilage do not inevitably lead to DJD but are considered to be one of the driving factors in the development of osteoarthritis, a syndrome characterized by chronic progressive structural damage in the joint. 99,100,144 Morphologic changes in the aging articular cartilage are similar for humans and animals. Usually, the major weightbearing joints are involved, meaning that the anatomic localization differs between species. Despite morphologic similarity, different terminology has been used in human and veterinary literature. In human medicine, DJD is often designated as osteoarthritis (OA), whereas in veterinary medicine, the term degenerative joint disease is preferred, based on the pathogenesis. Aging in the articular cartilage of humans and animals is characterized by changes in both matrix components and cellular components. 99,144 Loss of matrix proteoglycans and aggrecans results in exposure of collagen fibrils with roughening of the surface (fibrillation), which causes decreased stiffness and increased vulnerability to damage. 99 Also, collagen fibrils of the cartilage thicken and develop increased cross-linking. 99 On the cellular level, the chondrocyte density decreases with a decline in mitotic activity, increased senescence, and decreased synthetic activity. 99,100,144 Aged cartilage with weakened mechanical properties can subsequently develop fissures in the superficial cartilage that may extend into the deeper layers with progressive loss of cartilage (ulcerations) and secondary involvement of the subchondral bone (eburnation). 144
Several animal models for human DJD have been used, each with its own advantages and disadvantages (eg, mouse, rat, guinea pig, rabbit, dog, sheep, and horse). 103,144 The role of mouse models in OA/DJD research is increasing because of the availability of genetic manipulation in this species. 47 The most appropriate joint to select for OA research in mice is the knee (femorotibial) joint. Spontaneous development of OA/DJD has been reported in the knee joint of C57Bl/6 mice at the age of ∼17 months. 47,105 Furthermore, obesity in mice is associated with a marked increase in the severity of the knee joint lesions, similar to the correlation between obesity and OA in humans. 47 Recently, McNulty et al 104,105 published a new grading scheme for the histopathology of OA in the knee joint of mice. This grading scheme is aimed at identifying lesions in multiple tissues (eg, articular cartilage, meniscus, subchondral bone). Previous grading schemes focused primarily on lesions in the articular cartilage but appeared to have low sensitivity and reproducibility in mice. 104 The lack of reliability of these older grading schemes is likely due to the variable ability to identify the different zones in the very thin articular cartilage of mice. 105
Published data on DJD in progeroid mouse models are limited. In-depth analysis of knee-joint lesions has been published for XpdTTD /XpdTTD mice, a progeroid mouse model for human trichothiodystrophy. 19 The results of this study demonstrate the highly segmental nature of progeroid syndromes: Although these mice had prominent osteoporosis, the age-related lesions in the articular cartilage were not more severe compared to wild type mice, and female XpdTTD /XpdTTD mice were even partially protected from cartilage damage. 19 Both wild type mice and male XpdTTD /XpdTTD mice showed superficial fibrillation with occasionally small fissures in the articular cartilage of the lateral side of the tibial plateau. 19 These lesions were mild at 52 weeks of age and progressed to moderate severity at 104 weeks of age. 19 The female XpdTTD /XpdTTD mice showed less cartilage damage and less loss of articular cartilage proteoglycans at 104 weeks of age. Although there was no increased severity of the age-related articular cartilage lesions in XpdTTD /XpdTTD mice when compared to wild type mice, XpdTTD /XpdTTD mice did show a marked decline in the thickness of the subchondral bone plate that was not observed in wild type mice. 19
Loss of articular cartilage with subchondral bone sclerosis and ankylosis in the knee joint has been reported in adult Gsk3α -/- mice as part of a progeroid syndrome with concurrent phenotypes in other organs. 170 These mice also developed vacuolar degeneration and atrophy of the skeletal muscle, which might have led to joint instability and therefore potentially contributed to the severity of the joint lesions.
Intervertebral disc degeneration is one of the most common debilitating diseases in the elderly population. 134,152 Aging is considered to be one of the major predisposing factors for the development of disc degeneration, together with genetic predisposition and mechanical factors. 1,152 Changes that occur in the intervertebral disc with aging are a decrease of the number of notochordal cells and the amount of proteoglycans in the nucleus pulposus, with loss of hydration and viscosity. This leads to condensation and fragmentation of the nucleus pulposus with loss of the turgor that supports the annulus fibrosis. This allows the annulus to bulge outward, making it more vulnerable to tears and damage, starting a detrimental cycle of progressive disc degeneration. 1 Reduced cell number in the annulus fibrosis contributes to lower collagen turnover with increased cross-linkage and thicker, stiffer collagen bundles. In addition, glycosylation of collagen fibers generates even more cross-links, contributing to stiffening of the annulus and the macroscopic yellow appearance of aged discs. 1,2
Among domestic animals, spontaneous intervertebral disc degeneration is most frequently described in dogs. Some authors suggest that dogs with spontaneously occurring intervertebral disc disease could be an appropriate model to study human intervertebral disc degeneration. 13 Other animals with spontaneous disc degeneration are the sand rat, pintail mouse, Chinese hamster, and baboon. 134 A major disadvantage of these models is the complex and multifactorial basis of disc degeneration, which makes it particularly difficult to identify potential molecular mechanisms. This complicating multifactorial basis can be partially avoided by using progeroid mouse models. 152 Furthermore, the intervertebral lumbar discs of mice share many geometric characteristics with human intervertebral discs (eg, disc height, anteroposterior width, nucleus pulposus area). 113 Whereas some progeroid mouse models display intervertebral disc degeneration, to the best of our knowledge, there are no reports specifically describing intervertebral disc degeneration as being part of the major human progeroid syndromes (ie, Hutchinson-Gilford progeria, Werner syndrome, and Cockayne syndrome).
Among progeroid mouse models, intervertebral disc degeneration is probably best characterized in the Ercc1 -/Δ mouse model. Intervertebral disc degeneration in these mice mimics age-related disc degeneration in aging animals. 108,152,153 Phenotypes were observed in mice as young as 8 weeks and developed progressively with an aggravated phenotype in 20-week-old mice that resembled the appearance of aged intervertebral discs in wild type mice of 2 years and older. 153 Accelerated intervertebral disc degeneration in these mice is macroscopically characterized by loss of the gelatinous appearance of the nucleus pulposus and increased amounts of fibrous tissue. 153 Reduced thickness of the intervertebral discs has also been described but was measured using microcomputed tomography and was not typically noted on macroscopic evaluation. 153 Histologically, these intervertebral discs were characterized by a decreased cellularity, particularly near the end plate. 153 Reduced amounts of proteoglycans in the nucleus pulposus were demonstrated with safranin O staining. 108,153 These changes were associated with increased numbers of p16-expressing senescent cells in the nucleus pulposus and increased numbers of apoptotic cells in the annulus fibrosus as demonstrated with TUNEL staining. 108,153 The observations made in these DNA repair-deficient Ercc1 -/Δ mice suggest that DNA damage plays an important role in disc aging and intervertebral disc degeneration.
Kyphosis has an estimated incidence in older humans of 20% to 40%. 77 The underlying causes and contributing factors of kyphosis in elderly people have not been fully elucidated and cannot be completely explained by osteoporosis. 77 Changes in muscle strength, ligaments, intervertebral discs, and bone density are all thought to be involved in this syndrome. 77 The combination of osteoporosis, intervertebral disc degeneration, weakening of ligaments, and muscle atrophy may also be responsible for the development of lordokyphosis in multiple progeroid mouse models. Thoracic lordokyphosis in these mice is often reported as a macroscopic observation. Development of lordokyphosis has been reported to start as early as 10 to 11 weeks of age for Xpg -/- mice, 25 weeks for PolgAmut /PolgAmut mice, and 31 to 33 weeks for Ku80 -/- Rag-1 -/- mice and Ku80 -/- mice. 11,69,146,154 Multiple authors attribute the presence of lordokyphosis to vertebral osteoporosis, although in many of these studies, analysis of the skeletal muscles, intervertebral discs, and ligaments is not included. It is interesting that Wijshake et al 160 attribute early development of lordokyphosis to early muscular atrophy in Bub1b +/GTTA mice, since these mice did not show any evidence for reduced bone density. This raises the question as to which extent muscular atrophy might play a role in the other mouse models with kyphosis. These findings support the assumption that kyphosis is a multifactorial senile phenotype that results from a combination of simultaneous age-related lesions such as osteoporosis, intervertebral disc degeneration, and muscular atrophy. Therefore, a multimodal approach for analyzing complex phenotypes like these will reduce the risk of misinterpretation of the observations made in tissues that are not involved or only partially involved in the development of the phenotype. Microcomputed tomography and magnetic resonance imaging (MRI) are gaining popularity in animal studies on joint diseases and these techniques complement histopathology. 144 Microcomputed tomography allows high resolution 3-dimensional imaging of bone architectural changes that cannot be obtained with histopathology but has low soft tissue contrast. 144 In addition, microcomputed tomography has the advantage of being noninvasive, allowing longitudinal follow-up of the disease progression. 144 Micro-MRI, which is more suitable for imaging soft tissues, has been recently developed for rodents but needs further improvements for in vivo use. 144 Both methods can guide the histopathologic analysis by indicating which tissues and anatomic areas are of interest for further histopathologic examination.
Skeletal Muscle
Aging is inevitably accompanied by loss of muscle mass, known as sarcopenia or senile muscle atrophy (Table 5). 49,101,126 In the elderly population, senile sarcopenia is considered responsible for frailty, higher risk for falling accidents, disability, and even mortality. 49,61,95,101 Respiratory infections are common in the elderly population, and atrophy of the respiratory muscles (eg, diaphragm) may be a predisposing factor for this potentially fatal condition. 61 Loss of muscle mass with aging is a gradual process, in humans starting at age 50 and progressing to 30% to 50% loss of muscle mass by the age of 80. 5,24,49 Similar observations have been made in rodents, although compared to humans, the percentage of muscle fibers that are lost over a comparable lifespan period is smaller. 49
Sarcopenia is complex in its pathogenesis. Although loss of motor units is considered to be an important underlying cause, 49,95 there are multiple other potential mechanisms involved such as myocyte apoptosis, altered protein synthesis and turnover, as well as impaired satellite cell function. 24,95 Studying sarcopenia involves antemortem evaluation of physiologic and functional parameters such as physical activity, endurance, grip strength, and whole body tension. Macroscopic examination of sarcopenic mice will reveal reduced body weight, visible reduction in muscle mass, and reduced muscle weight. Histologically, sarcopenia is characterized by both loss of muscle fibers and a smaller fiber cross-sectional area, defined as fiber atrophy. 44 Furthermore, there is transition of “fast” type II fibers to “slow” type I fibers, 40,126 and variable replacement of myocytes by connective tissue or fat infiltration is possible. 44 Other less reported aging phenotypes in skeletal muscle are tubular aggregates in the type II myofibers of aged, inbred male C57BL/6, BALB/c, DBA/2, 129 Sv, and 129Ola mice and intracellular lipofuscin accumulation. 3,34 Tubular aggregates are composed of accumulated stacks of sarcoplasmic reticulum and can be visualized by electron microscopy or histochemical staining with modified Gomori trichrome staining, on which they appear as bright red inclusions. 34,130 In humans, tubular aggregates are considered a feature of myopathies and associated with a variety of clinical syndromes; this is in contrast to male inbred mice (C57BL/6, BALB/c, DBA/2, 129 Sv, and 129Ola strains), in which tubular aggregates are frequently observed and not necessarily associated with muscle pathology. 34,130
Sarcopenia has also been observed in several progeroid mouse models, and histologic examination reveals slightly different characteristics, depending on the mouse model. 126 Zmpste24-/- mice showed an altered fiber size distribution and increased numbers of myocyte nuclei. 60 Although the collagen content relative to muscle mass was elevated when measured with biochemical assays, on histology there was no significant fibrosis. 60 In the author’s opinion, this may be explained by the reduction in size and volume of the myofibers, which leads to a condensation of interstitial connective tissue in the muscle and thereby alters the collagen mass per muscle mass unit. Ku80 -/- mice exhibit a marked decrease in myofiber size with a shift in fiber type toward a type I dominated phenotype and paradoxically increased regenerative capacity after single event muscular damage. 40 In addition, these mice show other signs of progeria, poor postnatal growth, and smaller body size. 40 In contrast, heterozygous Ku80 +/- mice develop a more modest reduction in myofiber size that is also associated with an age-associated fiber type shift, but these mice grow normally and do not develop other signs of progeria. 40 The regenerative capacity of the skeletal muscle in 2-month-old Ku80 +/- mice was reduced, characterized by increased interstitial fibrosis with adipocyte infiltration and an increased number of infiltrating cells (not further specified), similar to the impaired regenerative response observed in 18-month-old wild type mice. 40 These results demonstrate that heterozygous Ku80 +/- mice share more similarities with physiologic aging than Ku80 -/- mice and might be a more appropriate model to study sarcopenia. 40
Other progeroid mouse models that are known to develop accelerated sarcopenia are Bub1bH/H and Bub1b+/GTTA mice. 9,10,160 In these mice, average muscle fiber diameter was reduced in gastrocnemius, paraspinal, and abdominal muscles, with increased myofiber size variation, intermuscular fibrosis, and impaired regenerative capacity. 10,160 Apoptosis and senescence could contribute to the observed muscle atrophy in this model as the transcript levels of p16 and p19 were elevated in the skeletal muscle of Bub1b+/GTTA mice. 160 Cellular senescence is proposed as a major contributing factor in skeletal muscle aging and impaired regenerative response as inactivation of p16 restores the regenerative response and attenuates premature aging in the skeletal muscle of Bub1bH/H mice. 10 Sarcopenia has also been reported as one of the lesions in the progeroid phenotype of Ercc1 -/- mice. 110
Whereas the models discussed so far display more or less similar features of skeletal muscle aging, another age-related phenotype has been reported in Gsk3α-/- mice. These mice have a shortened lifespan and develop marked sarcopenia in both skeletal muscle and heart, histologically characterized by vacuolar degeneration of myofibers and intracytoplasmic tubular aggregates in the skeletal muscle myofibers. 170 Tubular aggregates have also been demonstrated in male SAMP8 mice, 111 a senescence-accelerated mouse model that also displays decreased fiber size and a numeric shift toward type I fibers. 126 The significance and role of tubular aggregates is not fully elucidated. Appearance of tubular aggregates has been suggested to relate to androgen and changes in calcium homeostasis 34 and more recently to protein accumulation, potentially as part of physiologic aging. 130 Interpretation and translation of this observation from mouse models to human medicine should be done with extreme caution. The clinicopathologic features of tubular aggregates seem to be different for mice and humans, and their role in muscle aging is still unclear. 34
Nervous System
Brain is probably the most vulnerable tissue affected by aging due to its high oxygen requirements, limited regenerative ability, and low rate of endogenous antioxidant. 42 Brain aging is associated not only with pathologic morphologic phenotypes but also with multiple cognitive deficits and behavioral abnormalities. Common brain aging diseases include Alzheimer’s disease of humans and cognitive dysfunction syndromes of dogs and cats. In humans, common age-related neurologic lesions include brain atrophy, neuronal loss, neuropilar deposition of amyloid beta or senile plaques, intraneuronal taupathies (neurofibrillary tangles or NFT), cerebrovascular amyloid angiopathy, neuronal lipofuscinosis, vascular and meningeal mineralization, loss of white matter integrity, and astrogliosis. 133 All of these lesions except NFT were also reported in aged domestic animals and nonhuman primates. Age-related brain atrophy, gliosis, neuronal loss, and neuronal lipofuscinosis were reported in wild type mice and rats. 15,66,138
Neurodegenerative changes develop at an early age in the brain of several progeroid mouse models (Table 5). Bub1bH/H mice develop increased numbers of astroglia and microglia at the age of 1 month and 5 months, respectively, resembling age-associated gliosis. 66 Xpg -/- mice have been reported to develop astrogliosis with increased microglial activation in brain and spinal cord, starting at 4 weeks of age and progressing over the next 10 weeks. 11 At 14 weeks of age, astrogliosis was severe and associated with degeneration and loss of cerebellar Purkinje cells, axonal spheroids, and torpedoes and increased numbers of apoptotic cells throughout the brain. 11 Degeneration and loss of Purkinje cells, however, do not resemble a typical age-associated lesion but are more similar to age-related neurodegenerative disease. Neurodegenerative lesions similar to those of Xpg -/- mice have been demonstrated in the spinal cord of Ercc1 -/Δ mice. 39 In addition to the neurodegenerative lesions in the central nervous system, these Ercc1 -/Δ mice displayed age-dependent motor neuron loss with neurofilament accumulation in the distal motor axons and denervation atrophy of skeletal muscles. 39 Peripheral neurodegenerative lesions were also demonstrated in the sciatic nerve of Ercc1 -/Δ mice. 58 At 8 weeks of age, the nerve fascicles were 20% smaller than those of the littermate controls but were normally organized. 58 Rapid progression of the neurodegenerative phenotype occurred. At 20 weeks of age, the fascicles were not only smaller but also disorganized with loss of large nerve fibers, a larger percentage of small nerve fibers, loss of myelin, and an increased amount of endoneural connective tissue. 58 The ultrastructural features of this phenotype indicate axonal atrophy with secondary myelin degeneration (ie, redundant myelin, crenated myelin sheaths, paranodal loops of myelin, and myelin droplets and ovoids). 58
Several transgenic mouse models were generated to mimic the pathologic and cognitive phenotypes of AD and other human neurodegenerative disease such as Parkinson’s disease. More information on spontaneous age-related neurologic lesions in rodents and the available progeroid mouse models of neurodegenerative disease can be found in this special issue in the review by Youssef et al. 168
Skin
An important reason to study cutaneous aging lies in the reduced wound healing ability of the aged skin, causing nonhealing wounds and ulcers in the elderly population that can lead to severe infections 57,167 and increasing the risk of wound dehiscence after surgical procedures. 57 In human skin, 2 types of aging are recognized: intrinsic aging, which develops independently of ultraviolet (UV) light exposure, and extrinsic aging, also known as premature skin aging or photoaging, driven by UV light-induced damage. Similar molecular mechanisms are involved in both types of aging. 82 Changes in human aging skin include atrophy, drying, roughness, altered pigmentation, wrinkling, and an increased incidence of neoplasms as well as hair graying and hair thinning or loss (alopecia). 57 Similar changes are part of human progeroid syndromes like Hutchinson-Gilford progeria, Werner syndrome, Rothmund-Thomson syndrome, and Cockayne syndrome. 67 Histologically, most changes occur in the dermis and consist of decreased dermal thickness and loss of dermal-epidermal interdigitations, indicating atrophy. The collagen content and cellular content (ie, fibroblasts, mast cells, and macrophages) decrease. Collagen and elastin bundles become less organized. 48,57,82 Furthermore, dermal blood vessel size and density decrease with age. 82 Epidermal changes are less prominent and consist of loss of epidermal rete pegs, progressive reduction in the number of melanocytes, and decreased numbers of Langerhans cells. 48
Age-related changes in the skin of animals are rarely described, but regional graying of hair and skin atrophy are known to occur in many domestic animals and also in mice. When using animal models to study aging changes in the skin, one should be aware that most domestic animals have a straight dermal-epidermal junction; this is a normal finding that should not be interpreted as age-related loss of the interdigitations (rete pegs) that occurs in humans. 55 In mice, interdigitations between the dermis and epidermis are present only in the soles of the feet. 139 Other differences between the human skin and mouse skin involve the hair follicles and pigmentation. In human skin, individual hair follicles cycle independently, whereas in mice, hair follicles cycle in waves. 139 Melanocytes are differently distributed. In human skin, melanocytes are present in both the epidermis and hair follicles. In mice, melanocytes are restricted to the hair follicles in the haired skin and epidermal melanocytes occur only in the nonhaired or sparsely haired skin of the tail, feet, and ears. 137
Age-associated atrophy of the skin is often poorly defined in research papers. The term is most commonly used to indicate a reduced thickness of the dermis, associated with altered collagen organization, reduced cellularity within the collagenous connective tissue, and reduced amounts or absence of the subcutaneous adipose tissue. Some reports include thinning of the epidermis and atrophy of the panniculus carnosus muscle. Skin atrophy has been reported for multiple progeroid mouse models, including Col1α2-Cre +/- Sod2 -/f mice, 145 LmnaL530P/L530P mice and LmnaG609G/G609G mice, 106,115 Klotho-/- mice, 167 Bub1bH/H mice, 9 Ku80 -/- mice, 69,154 and Dna-pkcs −/− mice 123 (Fig. 2, Table 6).
Pathologic phenotypes in the skin, immune system and eye of progeroid mouse models.

aNomenclature rules: see footnote of Table 4. SAMP (senescence accelerated prone mouse) indicates a mouse strain, not a gene.
bLymphoid depletion was detected in different organs for different progeroid mouse models, including spleen, thymus, and bone marrow. Xrcc5 -/- mice displayed increased apoptosis in the spleen and lymphoid tissues. 169
Wound healing in elderly people is characterized by delayed onset of granulation tissue formation, re-epithelialization, angiogenesis, and collagen synthesis, when compared to wound healing in young people. 57 Impaired macrophage function is considered to play a major role in age-related repair defects, but reduced proliferative capacity of keratinocytes, fibroblasts, and vascular endothelial cells likely contributes. 57 We recently demonstrated that senescent fibroblasts and endothelial cells within the wound gap are also essential for optimal wound healing through secretion of PDGF-AA. 38
A potentially useful mouse model to study the aging skin is Klotho -/- mice, which display premature aging (eg, osteoporosis, skin atrophy, infertility, atherosclerosis) starting from 3 to 4 weeks of age and die at 8 to 9 weeks (Table 2). 167 The skin of Klotho -/- mice is characterized by reduced numbers of hair follicles, reduced epidermal and dermal thickness, and absence of subcutaneous adipose tissue, similar to the morphologic appearance of senile human skin. 167 Yamashita et al 167 created surgical wounds of 8 mm in diameter in the dorsal skin of Klotho -/- mice and wild type mice. Klotho -/- mice showed enlargement of the wound in the first 5 days and lack of wound contracture, followed by healing with complete closure at 35 days. 167 In contrast, wild type mice showed much faster healing (complete healing in 19 days) with wound contraction and no initial enlargement of the wound. 167 Histologically, granulation tissue formation, collagen deposition, and infiltration of inflammatory cells were less in 4- and 7-day-old wounds of Klotho -/- mice compared to wild type mice. 167 Klotho -/- mice could be useful to study age-related skin atrophy and delayed wound healing; however, one disadvantage of this mouse model is the high rate of unexpected death of these animals when exposed to stress (eg, transportation, physical activity). Mortality rates can be as high as 50%, complicating relatively long-term follow-up of these mice. 167 Another disadvantage could be the debatable translational value of Klotho mice as a model for aging. Recent evidence demonstrates that expression of Klotho in the kidney is primarily responsible for the systemic Klotho-mediated effects. 92 In mice, nephron-specific Klotho deletion results in a phenotype that is identical to systemic Klotho deletion. The observations from this study imply that chronic kidney disease results in a state of KLOTHO deficiency. 92 Based on this information, Klotho -/- mice could be regarded as a model of chronic renal failure, rather than a model of aging. Furthermore, other authors have stated that the marked hyperphosphatemia that occurs in Klotho -/- mice is responsible for the aging phenotype, as low phosphate diets rescued both vascular lesions and the shortened lifespan in these mice. 85 Delayed wound healing has also been reported in Bub1bH/H and Terc -/- progeroid mouse models. 9,127
Age-associated hair loss and graying has been investigated in a remarkable number of research papers, which focus on the mechanisms and the role of alterations in the stem cell niche in the development of these phenotypes. Although graying of hair has been reported to occur in progeroid telomerase-deficient mice, in general, progeroid mouse models evaluated for this review have not been used to study the mechanisms involved in graying. 127
Age-associated alopecia is another phenomenon that occurs with aging in both mice and humans (Table 6). 32 In mice, the first hair cycle waves are highly synchronized, but with aging, they develop hair cycle domains. 119,131 Hair cycle domains are patches of skin with synchronized, coordinated hair cycling that cycle independent from adjacent hair cycle domains. 131 With increasing age, hair cycle domains get smaller and more fragmented, creating a complex pattern of smaller, nonsynchronized groups of hair follicles. 119 Furthermore, compared to young mice, old mice have a slower hair cycle with an expanded telogen phase. 32,131 Macroscopically, loss of fur is easily detected, and the histologic features of age-associated alopecia include an increased proportion of telogen hair follicles, 121 reduced follicular density, and follicular cellular atrophy. 54
The mechanisms for age-related hair loss and alopecia are not completely clear. It has been suggested that both reduced function and reduced number of epidermal stem cells in murine skin could contribute to the slowing of the hair cycle. 54 More recent, Chen et al 32 demonstrated that stem cell activating and inhibiting signals may play a more important role in age-associated alopecia. Since stem cells play a crucial role in both the normal and declining hair cycle, much emphasis has been on identification and quantification of these stem cells that are localized in the follicular bulge. The follicular bulge is prominent in the fetal skin but usually hard to morphologically distinguish in hematoxylin and eosin (HE)-stained sections of adult skin. 36,139 Furthermore, stem cells cannot be identified based on morphologic features in HE-stained sections; therefore, examination of the stem cell population in this area depends on the use of appropriate markers. Immunohistochemical markers that can be used to demonstrate epidermal stem cells in the bulge area are keratin 15, LIM homeobox protein 2, CD34, and α6-Integrin. 54 Furthermore, the gene markers Lrg5 and Lgr6 can be used to visualize stem cells in the follicular bulge; however, this requires additional genetic modification through integration of a reporter gene. 63,136
Multiple progeroid mouse models display early onset of 1 or more of the following skin phenotypes: alopecia, thinning of the skin, loss of subcutaneous fat, hair graying, delayed wound closure, and spontaneous wound development. These mutant mice include PolgAmut /PolgAmut , 146 Ercc1 -/-, 158 LmnaL530P/L530P and LmnaG609G/G609G , 106,115 Col1α2-Cre +/- Sod2 -/f , 145 Xpc -/- mice, 71 Tg(P16), 18 Bub1bH/H , 9 Tg(AIMP3), 114 Ku80 -/-, 69,123,154 and Terc -/-127 (Table 6).
Hematopoietic System
With aging, functional changes in the immune system lead to an increased susceptibility to infections, poorer response to vaccination, and increased prevalence of cancer. 29 Furthermore, many age-associated disorders such as atherosclerosis and Alzheimer’s disease are associated with a chronic low grade inflammatory state. 29 The functional changes associated with aging are most prominent in the adaptive immune system and lead to a decline in T and B cell function and reduced proliferative capacity of the bone marrow hematopoietic stem cells with a shift from lymphoid differentiation toward myeloid differentiation. 29,129 Age-related reduction in the number of T cells can be largely attributed to thymic involution. 29,155 Functional changes in the immune system are reflected by changes in the circulating cytokine profile, altered expression of CD markers, and changes in relative and absolute leukocyte cell counts. Morphologic studies of the aging hematopoietic and lymphoid system are few. An excellent review on age-related morphologic changes in lymph nodes is provided by Ahmadi et al. 4
Lymphoid depletion is observed in both aging humans and aging rodents and is characterized by progressive loss of cortical and paracortical lymphocytes, gradual disappearance of germinal centers, thinning of medullary cords, and expansion of sinuses. 4 Degenerative changes that may occur in lymph nodes of aged humans and mice include fibrosis, adipose tissue infiltration, and presence of fluid-filled cysts. 4 Fibrosis is characterized by deposition of hyaline, eosinophilic collagen and is also called hyalinization. 4 Lymph nodes from different draining areas can show these changes to a different extent but these variations seem inconsistent. 4 In general, the alterations that occur in other lymphoid and hematopoietic organs like the spleen, thymus, and bone marrow consist of reduction in lymphocytic cellularity, infiltration of adipose tissue, and fibrosis. 159 Senile amyloidosis may also occur in spleen and lymph nodes. In the aging thymus, reduced numbers of lymphocytes leads to the loss of morphologically distinctive cortex and medulla. 159 Age-associated decreased lymphocytolysis in the spleen and increased peribronchiolar lymphoid proliferation in the lung have been described in a lifespan study of aging in C57BL6/J mice. 76
Lymphoid depletion is also the most commonly reported change in the hematopoietic system of progeroid mouse models (Table 6). Examples are lymphoid depletion in the lymph nodes of Ku80 -/- mice (Fig. 3), depletion of the periarteriolar lymphoid sheaths in the spleen of p53 +/m progeroid mice, 147 thymic atrophy with reduced numbers of pre-B cells and thymocytes in Atm -/- mice, 165 bone marrow lymphoid depletion in Terc -/- mice, 128 accelerated involution of thymus and lymphoid depletion of the spleen in LmnaG609G/G609G mice, 115 increased apoptosis in the spleen and lymphoid tissues of Ku80 -/- mice, 169 and loss of bone marrow hematopoietic elements with adipose tissue replacement and accelerated thymic involution in Ercc1 -/Δ mutant mice. 41
Eye
With aging, structural changes in the eye lead to gradual loss of sight. Characteristic age-related changes are cataract, corneal endothelial degeneration, and retinal degeneration (Table 6). Of these changes, cataract has the highest incidence in aging humans. 20 Cataract also occurs in aging animals and is part of most human progeroid syndromes.
Cataract is probably the best-known ocular aging phenotype with incidences in the human population between 75 and 85 years of age as high as 45%. 46 Cataract development is strongly age related and an important cause of blindness in the elderly population. Currently, surgery is the only available therapy. 46
The incidence of cataract in wild type mice increases with age and has been reported to be 25% in CD-1 mice at the age of 18 months. 53 The morphologic characteristics of cataract are similar for many species including mice and humans. Macroscopically, cataract can be recognized by progressive opacification of the lens. Histologically, the changes can vary, depending on the stage of development. With typical cataract, the lens fibers will show swelling and vacuolation and Morgagnian globules may be present. Changes in the lens epithelium include hypertrophy with presence of large, vesicular bladder cells and fibrous metaplasia in which the epithelium becomes flattened and multilayered with extension of the lens epithelium over the posterior surface of the lens. 53 Advanced stages may exhibit dystrophic mineralization of lens fibers and liquefactive necrosis. Perilenticular inflammation can be present as a response to leakage of lens protein through the lens capsule (hypermature cataract). 53 Perilenticular inflammation may progress into panophthalmitis. 53
Cataract in the senescence-accelerated mouse (SAMP) has probably been best characterized from a morphologic point of view. Progressive cataract was evident from as early as 10 weeks of age and severity of the phenotype increased with age. 70 Mature cataracts in these mice were histologically characterized by posterior protrusion of the lens, rupture of the lens capsule, fibrous metaplasia of the lens epithelium, swelling of lens fibers, and liquefaction of the deep and perinuclear cortex. 70 Surrounding the area of the posterior lenticular protrusion was a funnel-shaped fibrovascular sheath containing the hyaloid artery. 70 These results indicate persistence and hyperplasia of the hyaloid vascular system. This developmental defect likely influences the development of cataract. So even though these mice display early onset and age-related cataract, it is probably not an appropriate model for human age-related cataract due to the interference of this developmental defect. Cataract has also been observed in progeroid Bub1b +/GTTA mice. 160 Lens changes included posterior migration of lens epithelium but Morgagnian globules were not observed. 160 In contrast, Bub1bH/H mice developed a more severe lens phenotype with cataracts characterized by the presence of Morgagnian globules. 9
The corneal endothelial layer fulfills important functions to maintain corneal transparency but is particularly vulnerable as these cells are not capable of regenerating after damage. 125 Loss of endothelial cells leads to irreversible corneal edema. With no pharmacologic therapies available, corneal transplantation is the only option to treat patients with corneal endothelial dystrophy. 125 Since loss of corneal endothelium is a gradual age-related phenomenon, most of these patients are older than 60 years. 125 Unmasking the underlying molecular pathways in the development of age-related corneal endothelial dystrophy could reveal potential targets for pharmacologic intervention. To the best of our knowledge, not much has been reported on spontaneous age-associated corneal endothelial dystrophy in aging rodents.
Progeroid Ercc1 -/Δ mice have been used as a model to study age-related corneal endothelial dystrophy. These mice develop early changes in the corneal endothelium, similar to the changes seen in old wild type mice. Histologically, there was increased apoptosis of corneal endothelium cells as demonstrated with TUNEL staining, with loss of endothelium, posterior projections extending from the corneal endothelium, and occasional adherence of CD45+ leukocytes. 125 On confocal microscopy, the corneal endothelium of Ercc1 -/Δ mice displayed early development of variation in cell shape (cellular polymorphism or pleomorphism), increased variation in cell size as defined by cell surface area (polymegathism), and presence of binucleated polyploid cells. 125 The term polymegathism specifically indicates size variation in corneal endothelium but almost invariably occurs together with other aspects of pleomorphism of these cells. Furthermore, cellular dropout and decreased cell density were observed with confocal microscopy. In addition, electron microscopy demonstrated thickening of Descemet’s membrane. 125
Retinal degenerative diseases are important causes of age-related vision loss. Of these, age-related macular degeneration is the most common, affecting approximately 8.7% of the population older than 50 years, and is influenced by both genetic and environmental factors. 28,150 Early changes in age-related macular degeneration are visible only on histologic examination and consist of linear and laminar deposits between the retinal pigmented epithelium (RPE) and Bruch’s membrane. 112 These deposits may act as precursors for drusen formation. Drusen, the clinical hallmark of age-related macular degeneration, are plaque-like extracellular aggregates composed of immune proteins, lipids, cholesterol, matrix proteins, and carbohydrates. 51,112 These aggregates are deposited between the basal lamina of the RPE and Bruch’s membrane and can be visualized clinically with fundus photography. 51,112 Drusen deposits are often associated with sub-RPE inflammation, neovascularization, and hemorrhages. 28,112 In late stage macular degeneration, there is geographic atrophy that can be observed with fundoscopy as sharply delineated patches of hypopigmentation in the central retina. 6,51 Histologically, geographic atrophy is characterized by focal loss of RPE, attenuation of choriocapillaris, Bruch’s membrane, and photoreceptor layer with or without choroidal neovascularization. 28,112 Although the disease name suggests lesions to be restricted to the macula lutea, it has been demonstrated that lesions also develop outside the macula. 112
An important difference between the human and mouse retina is the absence of a macula lutea in mice. Degenerative retinal changes are therefore expected to have a different distribution pattern in mice. In wild type mice, senile retinal atrophy may occur but incidence is highly dependent on the strain. 53 In mice, age-related retinal degeneration affects the outer segments of the peripheral retina. Other lesions of the retina and choroid are rare in mice. 53
In general, data on retinal changes in progeroid mouse models are rare. Currently, there is not a single mouse model that develops all the features of age-related macular degeneration. 51 Increased apoptosis in the retinal inner and outer nuclear layers, suggesting retinal degeneration, has been described for progeroid Xpg -/- mice. 11 SAMP8 mice show some of the changes that occur in human age-related macular degeneration (eg, atrophy of retinal pigmented epithelium and basal laminar deposits in the sub-RPE space) but do not develop the hallmark lesions such as drusen, choroidal neovascularization, and retinal atrophy. 97
Cardiovascular System
The risk for development of cardiovascular disease increases with age. Age-associated structural changes can lower the threshold for cardiovascular diseases such as atherosclerosis and diastolic heart failure and can contribute to disease severity. 86,122 Both in blood vessels and in the heart, age-associated structural changes can lead to increased stiffness, which can cause functional disturbances and development of cardiovascular disease. 86,162
Age-associated changes observed in human elastic arteries include aortic dilatation (chiefly in the ascending aorta) and marked thickening of the tunica intima that is most pronounced in the abdominal aorta. 151 Excessive thickening of the intimal layer can predict early, silent coronary artery disease (atherosclerosis) in humans; however, thickening of the intima is not necessarily associated with atherosclerosis. 86 Contributing to the increasing arterial wall thickness are sclerotic changes in the tunica media and adventitia, characterized by accumulation of collagen and disruption of elastin. 12 Medial smooth muscle cells reduce in number but enlarge and acquire different functional properties, shifting from contractile phenotype toward matrix-producing cells that are able to migrate into the intima. 12 Morphologic changes of the endothelium are not frequently described and include nuclear polyploidy and ultrastructural changes in the cytoskeleton. 12 Aging changes in vascular morphology are also frequently seen in the kidney. Renal arterioles show both intimal and medial thickening with normal aging; these changes can be accelerated and aggravated in hypertensive patients. 151 Age-associated changes in the vascular wall are largely due to changes in the relative amounts of cellular components (vascular smooth muscle) and matrix components (collagen, elastin). Stains that can discriminate these different components, like Masson trichrome and elastica von Gieson stain, are therefore helpful in histopathologic characterization of blood vessels. 102
Age-associated thickening of the arterial intima, similar to what has been described for humans, also occurs in rodents and nonhuman primates. Morphologic studies in rats demonstrated that the thickening of the tunica intima was due to increased amounts of collagen, fibronectin, and proteoglycans and proliferation of vascular smooth muscle cells. 88 Similar features are present in the regenerative response that occurs after vascular injury. 88 Furthermore, arteriosclerosis can develop in the arteries and arterioles of aged mice, most commonly in spleen, kidneys, and uterus; less frequently in heart, pancreas, and intestine; and only rarely in the large elastic arteries. 118 Not only can the tunica intima increase in thickness, but medial hypertrophy can also occur in the muscular arteries of aging mice. This can, however, be difficult to assess due to postmortem contraction of the arterial wall. 118 In contrast to humans, mice develop spontaneous atherosclerosis only at a very low incidence. 118
Vascular phenotypes have been described in progeroid LmnaG608G/G608G and LmnaG609G/G609G mice, Bub1bH/H mice, and SAMP8 mice (Table 7) but do not quite resemble the phenotype seen with natural aging. Lmna mutant mice are used as a model for Hutchinson-Gilford progeria syndrome (HGPS) in humans and show multiple signs of accelerated aging in different organ systems. Vascular changes in LmnaG608G/G608G and LmnaG609G/G609G mice are characterized by a progressive loss of vascular smooth muscle cells in the tunica media of the large arteries (including aortic arch), elastin fragmentation, and collagen and proteoglycan accumulation. 115,149 By 12 months of age, these mice showed nearly complete loss of vascular smooth muscle cells and replacement by fibrosis that extends into the surrounding adventitia, and by 16 months of age, calcification of the arterial wall was observed. Intimal thickening was minimal, in contrast to the phenotype of natural aging. 149 These important cardiovascular phenotypes were suggested to be responsible for the premature death of these mice. 115 Similarly, patients with HGPS develop accelerated aging phenotypes and premature, often fatal cardiovascular disease that is not considered part of the normal aging phenotype. 26 Due to the striking similarity between the vascular phenotype of this mouse model and that in HGPS patients, this mouse model is particularly useful for studying HGPS and seems less favorable for studying mechanisms involved in normal aging. Bub1bH/H mice are another progeroid model that develops a vascular phenotype. These mice show reduced numbers of vascular smooth muscle cells in the tunica media with thinning of the vascular wall; in addition, there was diffuse medial fibrosis and a decreased amount of elastin fibers. 102 The structural changes in the vascular wall that develop in some progeroid syndromes and in physiologic aging can predispose to development of atherosclerosis. This phenomenon was demonstrated in SAMP8 and SAMP1/Yag mice, which developed subendothelial fat deposits with infiltration of macrophages reminiscent of atherosclerosis. 50,166 Vascular calcification has been reported in Klotho -/- mice and Six2-Kl -/- mice (nephron-specific Klotho deletion) and will be discussed in the section on renal lesions. 75,85,92
Pathologic Phenotypes in the Cardiovascular System, Kidney, and Liver of Progeroid Mouse Models.
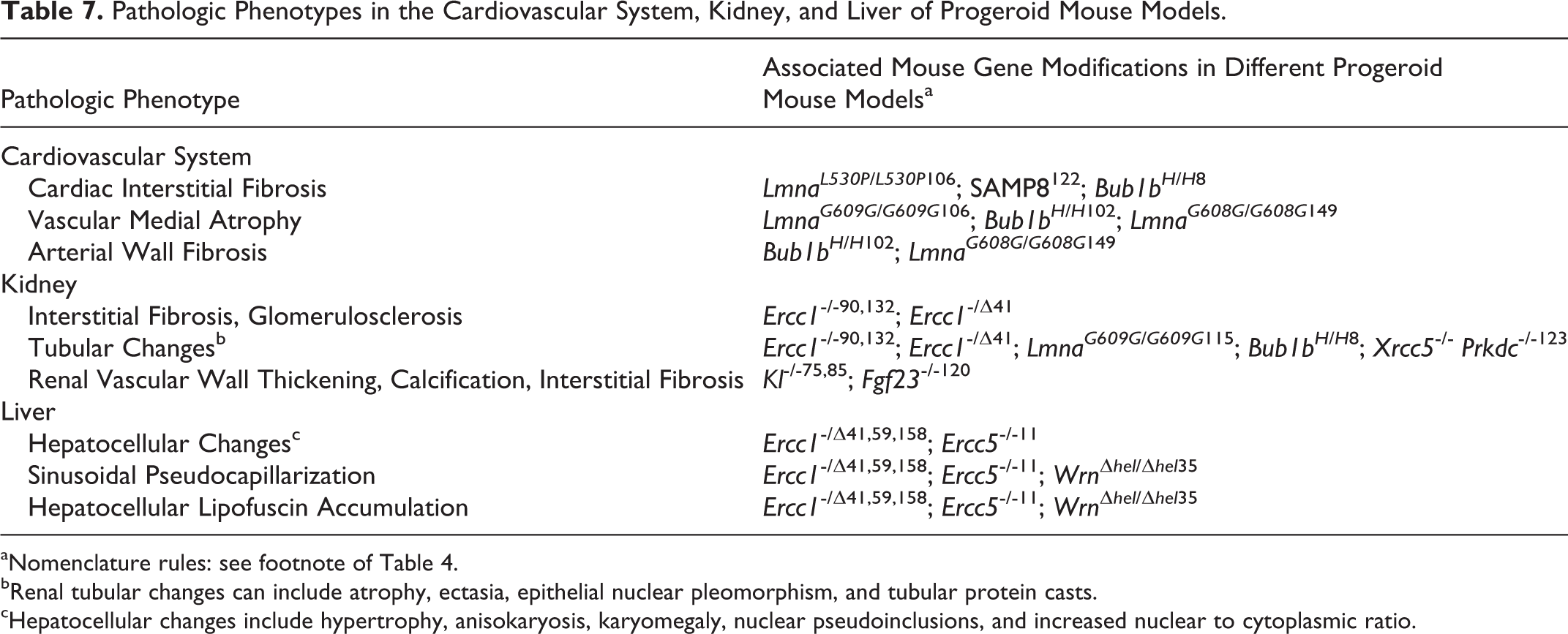
aNomenclature rules: see footnote of Table 4.
bRenal tubular changes can include atrophy, ectasia, epithelial nuclear pleomorphism, and tubular protein casts.
cHepatocellular changes include hypertrophy, anisokaryosis, karyomegaly, nuclear pseudoinclusions, and increased nuclear to cytoplasmic ratio.
The aging human heart shows progressive thickening of the left ventricular wall, and histologically, the cardiac myocytes are reduced in number but are larger due to hypertrophy. 87 Furthermore, there are areas of focally increased amounts of collagen, without alteration of the cardiac myocyte to collagen ratio. 87
Cardiac phenotypes in aging rodents are similar to those in humans and include thickening of the left ventricular wall, cardiac myocyte hypertrophy, and focal areas with increased amounts of collagen and reduced numbers of cardiac myocytes, which chiefly results from increased apoptosis. 88 Strain-specific background lesions that increase with age are spontaneous mineralization in the heart and myxomatous change of the subendocardial valvular interstitium. 141 One should be aware that these strain-specific background lesions can arise independently of the genetic disruption in a certain mouse model. Cardiac mineralization occurs in BALB/c, C3 H, and DBA mice with a strain-specific distribution pattern. BALB/c mice display mineralization with variable fibrosis in the epicardium of the right ventricular wall, C3 H mice show scattered mineralized foci throughout the entire myocardium, and DBA mice can show mineralization in both the epicardium and myocardium. 116 Spontaneous age-related endocardial myxomatous change has been reported in the heart valves of Swiss CD-1 mice, with a peak incidence at 700 to 738 days of age. 45 These lesions consist of nodular thickenings near the free edge of the valve. The nodules are composed of subendocardial fibromyxoid material that expands the interstitium. Pulmonic valves are most commonly affected. 45
In contrast to naturally aging rodents, which develop cardiac myocyte hypertrophy with aging, progeroid LmnaL530P/L530P homozygous mutant mice exhibited atrophy of cardiac myocytes associated with mild cardiac interstitial fibrosis characterized by increased numbers of fibroblasts per myocyte (Table 7). 106 Cardiac interstitial fibrosis has also been reported for SAMP8 mice and Bub1bH/H mice; both are progeroid mice that also display aging phenotypes in other organ systems. 8,122 In SAMP8 mice, the perivascular and interstitial cardiac fibrosis was associated with diastolic dysfunction, resembling age-associated diastolic dysfunction seen in elderly people. 122 Mild vacuolation of cardiac myocytes has been described for Ercc1 -/Δ mice, but this was a minor phenotype compared with the more marked phenotypes observed in other organ systems. 41 Dilated cardiomyopathy has been reported in progeroid Sox4 Δ/Δ mice. 52 Since SOX4 is involved in cardiac development, this phenotype might reflect a developmental disturbance, rather than a phenotype associated with accelerated aging.
Kidney
Aging of the kidney is a phenomenon that has been described for many species including humans and mice and is usually associated with a decline in renal function. Renal function is regarded as one of the major predictors of longevity and its loss influences the course of other diseases and the response to pharmacotherapy in the elderly population. 98 A wide variety of structural changes have been described for the aging kidney, as it is a complex organ. Sex and genetic background play a significant role in the development of these changes, in both humans and mice. 98 It is particularly difficult in the kidney to estimate the role of aging. 98 Many of the structural morphologic changes that are more frequently encountered with aging are also seen in renal diseases that are not related to aging. 98 Most of these structural changes predispose individuals to chronic kidney disease, characterized by loss of nephrons, progressive fibrosis, and a progressive decline in renal function. 75 As these changes often reflect an end-stage (eg, glomerulosclerosis), it is impossible to identify the inciting cause or contributing factors with certainty.
Macroscopically, human kidneys decrease in size starting at the age of 50 years, frequently associated with focal scarring. The incidence of renal cysts also increases with age. 98 Histologically, age-related structural changes can be seen in all renal components (ie, vasculature, glomeruli, tubules, and interstitium). Aging changes that develop in the human renal vasculature are similar to those that develop in the large systemic blood vessels, 98 including intimal and medial hypertrophy, fibrointimal hyperplasia, and arteriolosclerosis. 93,98 Similar changes occur in hypertensive disease and diabetes mellitus. 93,98 The vascular changes can eventually lead to glomerulosclerosis, a decrease in the number of glomeruli, tubular atrophy, and interstitial fibrosis, chiefly in the outer cortex. 98 Compensatory glomerular hypertrophy can develop in the juxtamedullary cortex. 98 Other glomeruli may show increased deposition of mesangial matrix and basement membrane. 98 Tubulointerstitial changes that occur with increased incidence with aging include tubular atrophy and loss, thickening of the tubular basement membranes, tubular ectasia, luminal eosinophilic hyaline casts, and interstitial fibrosis. 98 These changes contribute to macroscopically visible scarring and size reduction. On a cellular level, there is cytoplasmic lipofuscin accumulation within the proximal tubular epithelium, increased senescence of tubular epithelial cells, and increased numbers of apoptotic cells (not further specified). 98
Aging nephropathy, also called chronic progressive nephropathy, has been described for naturally aging mice and is morphologically similar to this entity in rats. 116,161 Early changes are in the cortex and outer medulla and chiefly involve the tubules. 161 Tubules with both epithelial degeneration and regeneration are recognized as separated foci of basophilic tubules and are surrounded by thickened basement membranes. 161 Proteinaceous casts are present within the medullary tubules. 116,161 In later stages, the areas with regenerative tubules enlarge and coalesce and show tubular ectasia. 161 Within the interstitium, there is infiltration of mononuclear inflammatory cells. With aging, there is also an increased incidence of glomerulopathy, likely due to immune-mediated glomerulonephritis. 116,141,161 These lesions start with proliferation and hypertrophy of mesangial cells, visible as clusters of irregularly shaped and sized mesangial nuclei surrounded by an increased amount of eosinophilic cytoplasm. 141,161 End-stage lesions include obliteration of the glomerulus with collapse of capillaries, deposits of hyaline eosinophilic material, and glomerular shrinkage (glomerulosclerosis) with adhesions between the glomerular tuft and Bowman’s capsule. 141,161 Often, these changes are associated with periglomerular fibrosis. 161 Occasionally, senile amyloidosis may involve the kidney, mostly within the interstitium of the cortex and corticomedullary junction. 161
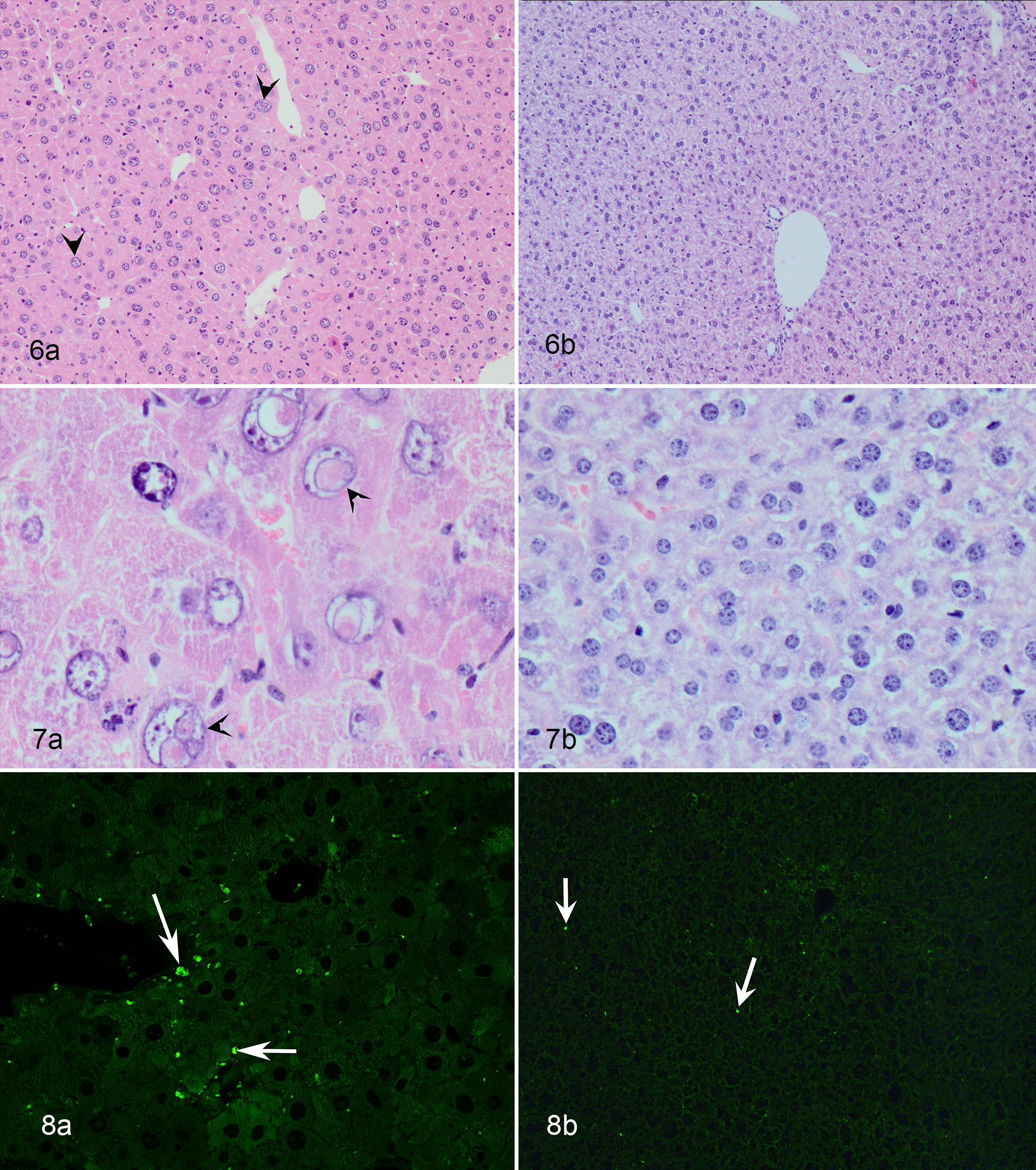
Several progeroid mouse models display a subset of previously described age-associated renal changes (Figs. 4–5, Table 7). Progeroid mouse models that develop changes in the kidney as a part of multiorgan involvement are Bub1bH/H mice, Ercc1 -/Δ mice, Ercc1 -/- mice, LmnaG609G/G609G mice, Ku80 -/-, Dna-pkcs -/-, Ku80 -/- Dna-pkcs -/- double knock out mice, and Klotho -/- mice. 8,41,75,90,115,123,132 Changes described for LmnaG609G/G609G mice are limited and consist of increased senescence-associated β-galactosidase staining in the renal tubular epithelium. 115 Progeroid Bub1bH/H mice show increased numbers of γ-H2AX foci, which indicate double-stranded DNA damage. However, overexpression of BUB1B showed the opposite effect, lowering the γ-H2AX expression and also decreasing the incidence of other common age-associated changes such as glomerulosclerosis, interstitial fibrosis, and tubular atrophy. 8 Changes in the Ku80 -/- and Dna-pkcs -/- mice include tubular epithelial anisokaryosis (polyploidy) and mild degenerative tubular changes with glomerular sparing. 123 Ercc1 -/- mice develop more extensive renal changes with segmental glomerulosclerosis, tubular ectasia with eosinophilic proteinaceous tubular casts, and epithelial attenuation (atrophy) and degeneration (Figs. 4–5). 90,132 Tubular loss and interstitial fibrosis have also been described. 90 Similar to polyploidy in the liver, the proximal tubular epithelium of Ercc1 -/- mice displays polyploidy characterized by marked nuclear pleomorphism, anisokaryosis, and occasional karyomegaly (Fig. 5). 132 Changes in nuclear morphology of renal tubular epithelium are not typically seen in aging wild type mice and could be a signature for defects in DNA repair mechanisms as is the case in Ercc1 -/- and Ku80 -/- and Dna-pkcs -/- mutant mice. 41
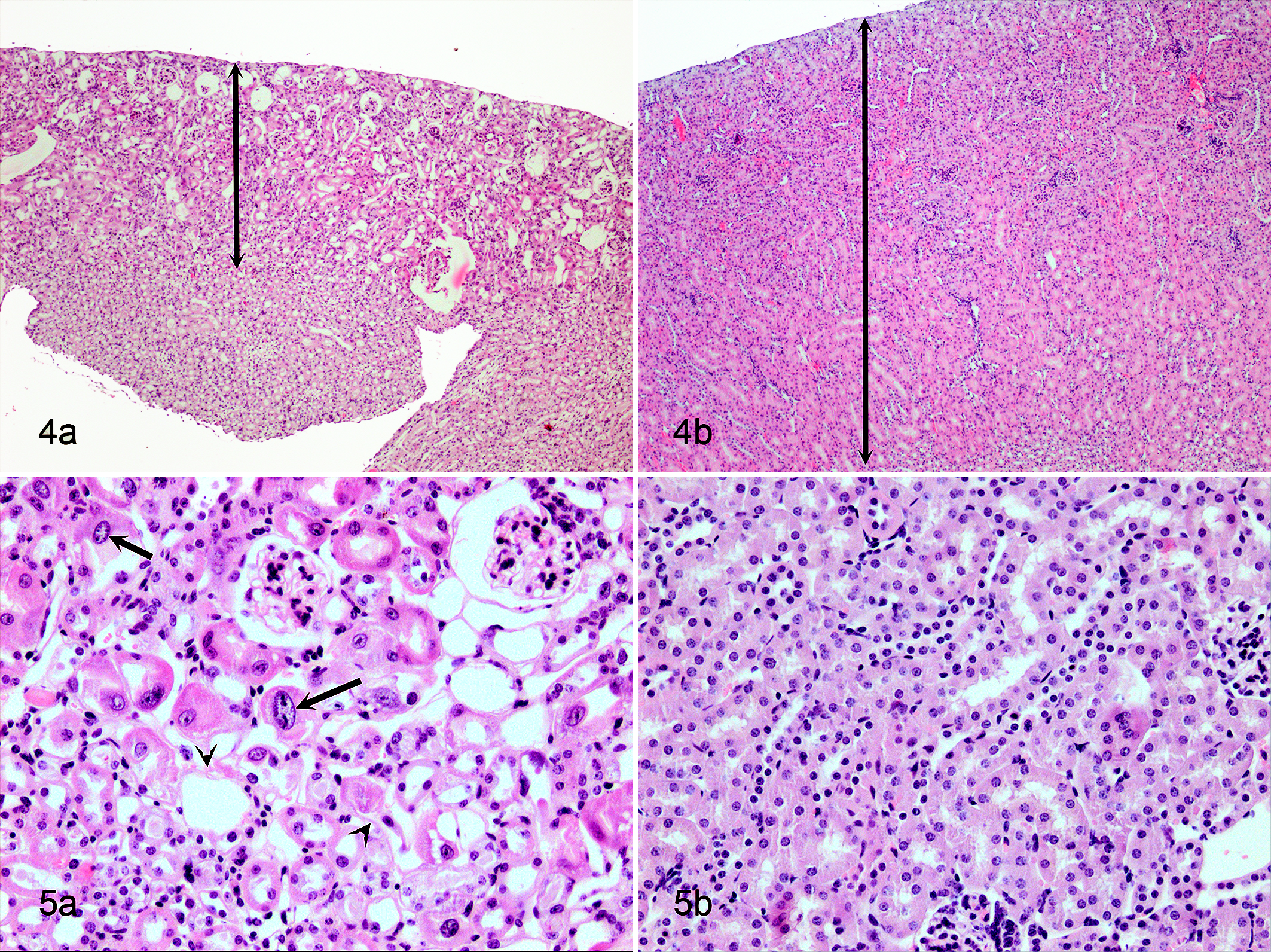
Renal aging has also been examined in Klotho -/- and Fgf23 -/- mice, but these mice exhibit renal changes that do not quite resemble the age-related renal phenotype in humans and mice. Renal changes that have been reported in Fgf23 -/- and Klotho -/- mice are widespread thickening of renal vessel walls with extensive vascular wall calcifications, tubular calcifications, and interstitial fibrosis. 75,85,89,92,120 In addition, these mice showed widespread ectopic calcifications in other soft tissues (eg, lung, skeletal muscle, skin, urinary bladder, testes, cardiac muscle). 75,89,120 Even though Klotho -/- and Fgf23 -/- mice develop changes that resemble natural aging (eg, kyphosis), the widespread calcification does not reflect normal aging but resembles a disease state that results from hypervitaminosis D, hypercalcinosis, and hyperphosphatemia. KLOTHO is required for FGF23 to function as a negative regulator of the phosphate balance. Both Fgf23 -/- mice and Klotho -/- mice develop hyperphosphatemia and exhibit similar aging phenotypes, which can be rescued by correction of the hyperphosphatemia. 75,85 Recently, Lindberg et al 92 demonstrated that the kidney is the principal source for circulating KLOTHO and responsible for Klotho-induced anti-aging effects. As discussed earlier in this article, Klotho -/- mice and Six2-Kl -/- mice (nephron-specific Klotho deletion) have identical phenotypes associated with KLOTHO deficiency and hyperphosphatemia. A comparable state of KLOTHO deficiency with hyperphosphatemia also occurs in chronic kidney disease. 92 These mouse models demonstrate that altered phosphorous metabolism may contribute to accelerated aging and is an important therapeutic target in patients with chronic renal disease. 85 Furthermore, this could indicate that chronic renal disease as a primary event induces secondary changes that resemble aging. 92
Liver
Diseases of the liver are also more prevalent in the elderly population and the aged liver is considered to have less capacity to adequately respond to a variety of injuries. 59 Aging in the liver is associated with multiple functional and structural changes of which the underlying mechanisms are not fully elucidated. 62 Macroscopically, the aging human liver decreases in size by 20% to 40% and often develops discoloration due to hepatocellular lipofuscin accumulation, known as “brown atrophy.” 7,62 This decrease in size has been attributed to a decrease in blood flow. 7 Histologically, one of the major changes in the human aging liver is pseudocapillarization, characterized by thickening of sinusoidal endothelial cells, decreased endothelial pores (fenestrations), and subendothelial perisinusoidal collagen deposition. 7,62 Pseudocapillarization of the sinusoids impairs exchange of nutrients, oxygen, and metabolites and thus impairs hepatocellular function. On the ultrastructural level, hepatocytes show a decrease in the amount of rough and smooth endoplasmic reticulum. 7,62 Together, these characteristics explain the altered drug metabolism and increased risk for adverse drug reactions in aged individuals. 62
Pseudocapillarization and lipofuscinosis are age-associated changes in the structure and morphology of the liver that are similar for humans and naturally aging mice. 59 Pseudocapillarization has been demonstrated in old mice and is characterized by increased thickness of the sinusoidal endothelium, increased numbers of stellate cells, and decreased porosity of the sinusoidal endothelium. 156 Most of these changes are best identified with electron microscopy, but thickening of the endothelium can also be identified on light microscopy, especially when using endothelial markers (eg, CD31 or factor VIII-related antigen). 59 However, collagen and basal lamina deposits in the perisinusoidal space could not be consistently demonstrated with immunohistochemistry for laminin and Masson trichrome stain and were limited to scattered patches on Sirius red staining. 156 In contrast, transmission electron microscopy allowed better demonstration of basal lamina and collagen deposition in the perisinusoidal space of older mice. 73,156 Other histologic changes in the murine aging liver include the presence of focal hepatocellular alteration, lipofuscinosis, hepatocellular hypertrophy, hepatocellular intranuclear pseudoinclusions, lipidosis, karyomegaly (polyploidy), and anisokaryosis and increased numbers of oval cells, stellate cells, and Kupffer cells. 7,59,62 Foci of hepatocellular alteration can be subclassified based on cellular tinctorial properties. 65,143 Morphology and incidence can differ among mouse strains; usually, the overall incidence is low and is higher in male than in female mice. 64,65 Predominant types of spontaneous age-related altered foci are basophilic, eosinophilic, and clear cell type. 65 Besides spontaneous occurrence with age, specific types of altered foci can also be induced by certain hepatocarcinogens. In mice treated with hepatocarcinogens, the incidence of altered foci is higher and these foci have been reported to precede neoplastic lesions, although their role in the development of neoplasia is not entirely clear. 64,65 These changes can be easily identified on light microscopy with HE stain. Lipofuscin pigment is a sensitive marker for aging in murine liver. This pigment is autofluorescent and can be easily scored and identified in nonstained paraffin slides using a fluorescent microscope. Furthermore, the incidence of background lesions such as hepatocellular necrosis, extramedullary hematopoiesis, mononuclear cell infiltration, random granulomas, and senile amyloidosis increases with age in the murine liver. 65 The occurrence of all of these aging changes depends on the mouse strain and can be influenced by environmental factors. Rare phenotypes in the aging mouse liver are sinusoidal dilation, angiectasia, bile duct proliferation, cholangiectasia, and cholangial cysts. 65
Progeroid Ercc1 -/Δ mice are probably the best characterized model with respect to accelerated aging of the liver (Figs. 6–8, Table 7). Gregg et al 59 concluded that the liver phenotype in Ercc1 -/Δ mice resembles that of naturally aged wild type mice and elderly humans. Ercc1 -/Δ mice developed early portal fibrosis and lipidosis, hepatocellular hypertrophy, karyomegaly, anisokaryosis, cytoplasmic nuclear pseudoinclusions (Figs. 6–7), increased nuclear to cytoplasmic ratio, lipofuscinosis (Fig. 8), increased numbers of stellate cells, and increased expression of CD31, which indicates enlargement of sinusoidal endothelial cells seen with pseudocapillarization. 41,59,158 Anisokaryosis and nuclear pseudoinclusions (cytoplasmic invagination into the nucleus and not true inclusions) are particularly prominent in the livers of progeroid and aging mice and caused by polyploidization. 41 Increased cellular senescence was demonstrated by increased β-galactosidase activity in both aged liver and the liver from progeroid Ercc1 -/Δ mice. 59 In addition, electron microscopy revealed thickening of the sinusoidal basement membrane and reduction in sinusoidal endothelial fenestration, supporting pseudocapillarization. 59 A liver phenotype similar to that of the Ercc1 -/Δ mice is present in the Xpg -/- mouse model. 11 Compared to Ercc1 -/Δ mice, the Xpg -/- mice have a shorter lifespan and develop similar aging phenotypes but slightly earlier. Another mouse model that is proposed as a model for human liver aging is the Wrn Δhel/Δhel mouse. 35 Pseudocapillarization similar to the phenotype in human aged livers was demonstrated in these mice, along with hepatocellular hypertrophy and increased amounts of hepatocellular lipofuscin. 35 However, other hallmarks of aging in the liver such as increased karyomegaly and anisokaryosis were not observed in these mice. 35 A different phenotype was described for Ku80 -/- mice. 154 The livers of these progeroid mice developed hyperplastic foci and the hepatocytes within these nodules showed signs of dedifferentiation. 154 In addition, these livers showed nuclear and cytoplasmic inclusions in the remaining hepatocytes, similar to what has been described for aging mice. 154
Gonads
Age-associated degeneration and atrophy of the gonads is a well-known phenomenon in both humans and animals 163,164 but is of minor importance in the field of geroscience. Macroscopic and histologic age-related changes are similar for mice and men. Macroscopically, aged atrophied testes are smaller and the parenchyma can be brownish due to lipofuscin accumulation. Histologically, the seminiferous tubules are reduced in diameter and show vacuolation and loss of seminiferous epithelium. 56,163,164 Spermatocytes, spermatids, and spermatozoa are the first to be affected, whereas Sertoli cells persist longest. 56 The epididymis also reduces in size due to atrophy of the tubular epithelium. 56 Lipofuscin accumulation occurs within Leydig cells and Sertoli cells and increases with age. Like the testes, the ovaries also atrophy with age. 96 This leads to a reduction in size and loss of all ovarian components. 96 Lipofuscin accumulation occurs within interstitial cells that form discrete islands. 96
Progeroid mouse models develop morphologically similar degenerative changes but at an earlier age. Not much emphasis is generally put on the description and characterization of the reproductive tract changes in progeroid mouse models. Few reports briefly mention the presence of testicular degeneration and/or atrophy and lipofuscin accumulation. 41,43,128,146,165 Even fewer reports mention changes in the ovaries, such as the absence of primary oocytes and follicles. 165 Age-associated changes in the male accessory sex glands and female reproductive tract have not been reported for progeroid mouse models to the best of our knowledge.
Intestinal Tract
Villous atrophy (reduced villus length) occurs in the proximal intestine of aging humans and mice; this potentially contributes to age-associated loss of body weight due to reduced nutrient absorption. 127 Among the literature available on progeroid mouse models, only a few mention morphologic changes in the gastrointestinal system. Accelerated occurrence of intestinal villous atrophy has been reported for telomerase-deficient Terc -/- mice, together with progeroid changes in other organs. 127,128
Discussion
In this review, we provide the most common macroscopic and histologic phenotypes of selected progeroid mouse models. Most of these mice develop a certain subset of age-associated lesions, thereby representing only some of the phenotypes of human aging, often referred to as segmental aging. Many of these models display morphologic changes similar to those that develop with normal aging and, therefore, could be of value for geroscience. The segmental character of the age-related morphologic changes implies that multiple models are needed to study the simultaneous changes that contribute to a certain age-associated phenotype in a particular tissue. This is exemplified by the different mouse models that are used to characterize senile osteoporosis. The different progeroid mouse models show that these models can closely mimic senile osteoporosis but may not be identical to the phenotypes of physiologic human aging. Different models have been used to demonstrate different characteristics of osteoporosis, suggesting that multiple underlying mechanisms are involved. Multiple mouse models may be needed to elucidate different parts of the highly complex events that occur in bone aging in humans. The same holds true for most other organ systems and tissues. The complex basis of aging is further supported by the fact that not all aging individuals develop all aging phenotypes. There is a large interindividual variability in the severity and spectrum of aging phenotypes, likely due to the heterogeneous genetic basis and contribution of environmental influences. Therefore, characterization of the pathophysiology of aging should include and combine data from multiple models.
Not all phenotypes encountered in progeroid mouse models resemble the changes that occur in natural aging. An example of this is Lmna mutant mice (LmnaL530P/L530P and LmnaG609G/G609G mice). These mice are used as a model for accelerated aging but the cardiovascular phenotype of these mice differs from the cardiovascular changes in natural aging of humans. In fact, more or less the opposite effect was observed: whereas with natural aging, the cardiac myocytes enlarge and arterial walls thicken, the Lmna-mutant mice demonstrated atrophy of cardiac myocytes and vascular smooth muscle depletion in the aortic arch. 106,115 Awareness of both similarities and differences between progeroid phenotypes and phenotypes of natural aging is important when these mouse models are employed in geroscience.
Currently, a large amount of data is available on progeroid mouse models. Many research papers have put emphasis on genetics and protein expression; pathology data are regularly included but often limited. In our opinion, considerable improvements could be made to obtain more information from these models. While reviewing research articles, we encountered several points for improvement.
First, when analyzing models of progeria, combining end-of-life studies with cross-sectional analysis can provide valuable information. This approach allows the differentiation of changes associated with “normal” aging from those of accelerated aging. Changes of normal aging are similar when comparing chronological age groups of wild type mice and mice with different genotypes. 41 Chronological age is calculated as the time point after birth. Biological age can differ from the chronological age and is based on the pathologic parameters and relative “fitness” or functional status of an individual in reference to chronological peers. 78 Changes that indicate accelerated aging are typically observed at an earlier chronological age, even if these changes develop at a comparable biological age relative to the total lifespan for that genotype. Thus, accelerated aging reflects a discrepancy between biological age and chronological age. Dollé et al 41 explained the advantages of combining cross-sectional analysis and end-of-life analysis. This approach allows for (1) certain individual endpoints, to characterize and differentiate between differences in aging rates (normal aging changes develop at a similar rate and are morphologically similar among wild type and progeroid mice in different chronological age groups), (2) characterization of age-associated changes at different time points on a biological scale (ie, relative to total lifespan), and (3) identification of age-associated pathology that is more severe on a biological age scale (“exaggerated aging”). 41 Full life-cycle studies are another method for more detailed phenotyping of age-related changes. These studies include mice from many different stages during life (ie, embryonic, fetal, infant stage, etc). This method is, however, quite expensive and therefore not very often used.
When analyzing tissues at different time points, organ-specific morphologic parameters that correlate with chronological aging could potentially serve as internal hallmarks of biological aging. For C57BL6 mice, this has been investigated in a limited set of organs and tissues. 76 Proposed parameters that correlate with chronological aging in these mice are lipofuscin accumulation in brain and liver, thickening of the glomerular membrane in the kidney, decreased lymphocytolysis in the spleen, and increased peribronchiolar lymphoid proliferation in the lung. 76 The use of these parameters as hallmarks of biological aging should be done with caution. The decreased lymphocytolysis and increased peribronchiolar lymphoid proliferation are not in line with the age-associated decrease in lymphoid cellularity that has been described by other authors. On an individual level, there is considerable interindividual variation in these parameters at a certain chronological age. That is, each individual has an individual rate of developing aging changes, and onset and rate of the development of aging changes can differ among organ systems. Thus, there is no consistent aging phenotype across multiple organs on an individual level, and the use of these parameters as biomarkers is limited to data analysis at the population level. 76 Furthermore, since tissues and organs have different rates of aging, exploration and validation of additional biomarkers as well as identifying different combinations of biomarkers are needed to find the most efficient combination of biomarkers to reflect overall aging. 78 Another point of concern is the differences among different mouse strains. Different mouse strains and substrains are known to display considerable variation in their physiology and behavior; therefore, it seems not unlikely that different mouse strains could also display variation within pathologic parameters of aging.
Considering the pathologic examination, most research papers do not explain the rationale for organ selection or exclusion nor do they routinely examine animals found dead or moribund. Some reports include only gross examination, which often does not provide insight into the underlying mechanism of phenotype development. Frequently, only a limited set of organs is sampled for pathology, and full necropsies were not reported in any of the studies consulted for this review. This could result in incomplete characterization of the phenotype due to lack of information on certain organs and tissues, and “unexpected” phenotypes could be missed. Also, it could influence the interpretation of the phenotype, as changes in 1 organ system can influence the development of changes in another organ or tissue. Performing full necropsies could reveal otherwise missed phenotypes and may contribute to a better understanding of the pathophysiology. There is a large amount of resources and references available that can support the pathology analysis of genetically engineered mice. An overview of the available resources is provided by Bolon et al. 16,17 A basic approach to perform pathologic examination on mutant mice is reviewed by Brayton et al. 21 An excellent, more detailed resource on full mouse necropsy procedures can be consulted online in the “Revised guides for organ sampling and trimming in rats and mice” (reni.item.fraunhofer.de/reni/trimming/index.php). The analysis of specific anatomic areas in certain tissues or the analysis of specific diseases may require a more advanced and multimodal approach, which depends on the research question. It is beyond the scope of this review to discuss all the different options regarding the more advanced and specific methods.
Another point for improvement is the use of proper terminology. Inappropriate terminology or incorrect terminology will lead to misinterpretation and a waste of data (eg, mineralization of periarticular tissue is not the same as osteophyte formation; it is not very informative to mention tumors without specifying a diagnosis or the organ or tissue affected). Providing a concise but correct pathologic description of the phenotype and including high quality images would support a better understanding of the phenotype among peers. Many papers include images that are either not in focus or too small to identify the described morphologic changes. Comparative pathologists can play a major role in improving the pathologic examination and presentation of the findings for progeroid mouse models. Comparative pathologists have been trained to use consistent and appropriate terminology, have profound knowledge of background lesions that could interfere with the interpretation of the phenotype, and understand the interrelationships of disease processes in different organ systems. 22,27,72
Clinical pathology data were rarely included in the reviewed research papers. Including clinical pathology data could be of interest in certain models to support the morphologic pathology data. Functional defects and metabolic derangements do not necessarily result in morphologic phenotypes but can have a major effect (eg, potassium levels influence cardiac rhythm, and hypokalemia can lead to muscular weakness).
In conclusion, progeroid mice are valuable models to study the mechanisms involved in the development of age-associated lesions. Their relatively simple genetic mutations provide a good starting point to correlate molecular pathways to morphologic and metabolic changes. When compared with normal aging mice, progeroid models have the advantage that they develop a predictable phenotype, in contrast to the more inconsistent development of the aging phenotype in wild type mice. A better knowledge of molecular pathways involved in development of the aging phenotype could expose targets for therapeutic intervention. However, translation of the data obtained from these models should be done with caution, as no model covers all the aging phenotypes and some models display changes that do not reflect the normal aging phenotype. Selection of the appropriate mouse model therefore requires well-founded knowledge of the phenotype. Pathologic examination can add valuable information, but state-of-the-art pathologic examination asks for a highly specialized, organ-specific approach beyond that described in this review. Awareness of the availability of highly sophisticated organ-specific analyses should drive researchers and pathologists to consult the appropriate resources and to collaborate with experts in the field of interest.
Footnotes
Acknowledgements
We thank Harry van Steeg and Martijn Dollé (RIVM, Bilthoven, The Netherlands), Jan Hoeijmakers (Erasmus MC, Rotterdam, The Netherlands), and Paul Hasty (University of Texas Health Science Center, San Antonio, Texas, USA) for kindly permitting us to publish images of their progeroid mouse models.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.