
Abstract
Mannheimia haemolytica serotype S1 is considered the predominant cause of bovine pneumonic pasteurellosis, or shipping fever. Various virulence factors allow M haemolytica to colonize the lungs and establish infection. These virulence factors include leukotoxin (LKT), lipopolysaccharide, adhesins, capsule, outer membrane proteins, and various proteases. The effects of LKT are species specific for ruminants, which stem from its unique interaction with the bovine β2 integrin receptor present on leukocytes. At low concentration, LKT can activate bovine leukocytes to undergo respiratory burst and degranulation and stimulate cytokine release from macrophages and histamine release from mast cells. At higher concentration, LKT induces formation of transmembrane pores and subsequent oncotic cell necrosis. The interaction of LKT with leukocytes is followed by activation of these leukocytes to undergo oxidative burst and release proinflammatory cytokines such as interleukins 1, 6, and 8 and tumor necrosis factor α. Tumor necrosis factor α and other proinflammatory cytokines contribute to the accumulation of leukocytes in the lung. Formation of transmembrane pores and subsequent cytolysis of activated leukocytes possibly cause leakage of products of respiratory burst and other inflammatory mediators into the surrounding pulmonary parenchyma and so give rise to fibrinous and necrotizing lobar pneumonia. The effects of LKT are enhanced by lipopolysaccharide, which is associated with the release of proinflammatory cytokines from the leukocytes, activation of complement and coagulation cascade, and cell cytolysis. Similarly, adhesins, capsule, outer membrane proteins, and proteases assist in pulmonary colonization, evasion of immune response, and establishment of the infection. This review focuses on the roles of these virulence factors in the pathogenesis of shipping fever.
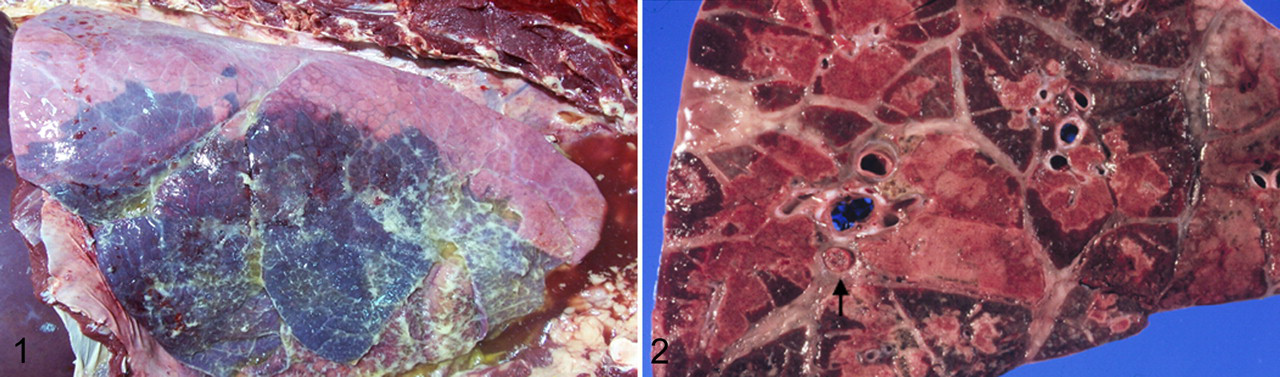
Mannheimia haemolytica (formerly Pasteurella haemolytica) is the major cause of fibrinous and necrotizing lobar pneumonia and pleuropneumonia of cattle which is often called shipping fever (SF) (Fig. 1 and 2). SF has a marked economic significance worldwide and costs approximately $1 billion per year to the US cattle industry. 125 The term shipping fever emphasizes circumstances (stress) under which the disease predominantly occurs. Various combinations of environmental stress can serve as cofactors in the pathogenesis of SF—such as inclement weather, shipment, weaning, overcrowding, and complex interactions among several infectious agents, including bovine herpesvirus 1, bovine parainfluenza virus 3, bovine respiratory syncytial virus, bovine viral diarrhea virus, Mycoplasma sp, and other bacteria. The underlying processes by which various environmental factors alone or in combination with microorganisms predispose cattle to SF are not fully understood. Alterations of local innate and adaptive immune responses presumably contribute to the development of the disease process. 45,64,107,109,110,111 Although SF is a multifactorial disease, M haemolytica serotype S1 is considered the major cause of the most severe form of SF pneumonia, and it is generally believed that control of M haemolytica S1 infection would markedly reduce the prevalence and severity of SF.
M haemolytica is an opportunistic pathogen; it is a normal inhabitant of the nasopharynx and tonsils of cattle and sheep. 46,118 In healthy cattle, the relatively nonpathogenic M haemolytica serotypes S2 and S4 predominate, whereas pathogenic serotype S1 is present in low numbers. 42,43 Although the exact mechanisms are not known, stress or concurrent viral infections result in microenvironmental change that favors increased multiplication and colonization of serotype S1 in the upper respiratory tract. 11 The selective growth and colonization of the nasopharyngeal region by M haemolytica serotype S1 is a prerequisite for the development of SF. 61 The resulting colonization of the nasal mucosa with large numbers of serotype S1 contributes to the inhalation of aerosol droplets containing bacteria, into the trachea and lungs. 50 Healthy calves can clear the inhaled bacteria; however, stressed calves develop pneumonia. Although the most common isolate from the cattle lung with SF is M haemolytica serotype S1; other reported serotypes include S2, S5, S6, and S9, and group S6 and S2 constitute approximately 25% and 10% of the isolates, respectively. 7,100
Various virulence factors possessed by M haemolytica S1 promote lung colonization and evasion of host immune response; therefore, the host immune status is critical in counteracting these strategies. When the host immune response is not successful, these interactions contribute to the development of SF. Multiple review articles have addressed these interactions in detail. 103,113 Several virulence factors of M haemolytica serotype S1 promote host–pathogen interaction and influence the outcome of infection. Of these, leukotoxin (LKT) and lipopolysaccharide (LPS) are important, and their roles in pathogenesis of SF are well documented. 23,61,98,113 Other virulence factors with less-defined pathogenic roles are capsule, adhesins, outer membrane proteins, and various proteases, such as neuraminidase and glycoprotease. 23,38,58,68 The following is an overview of the virulence factors of M haemolytica serotype S1.
Surface Proteins and Carbohydrates
Adhesins
A naïve host encounters M haemolytica from the environment through air or droplet inhalation. For development of SF, a necessary requirement is that of initial colonization and establishment in the nasopharynx by adherence to the epithelial surface. 17 This binding must provide resistance to the physical removal by air flow, local innate and adaptive immune mechanism, and mucociliary clearance. The adhesins of M haemolytica involved in the nasopharyngeal epithelial binding are not fully understood. 52 De la Mora et al 38 demonstrated a 68-kDa adhesin molecule associated with M haemolytica that is involved in the adhesion to cultured tracheal cells. In addition to binding tracheal cells, this adhesin binds a 165-kDa glycoprotein receptor onto neutrophils and initiates an oxidative burst. 38 Another putative adhesin molecule is present on fimbriae of M haemolytica and binds to a sialoglycoprotein receptor on respiratory epithelium. 56,88 However, a locus encoding such an adhesin has not yet been identified. 53 Kisiela and Czuprynski 63 recently identified the heat-modifiable M haemolytica outer membrane protein OmpA and lipoprotein Lpp1, which are involved in adherence to the bovine bronchial epithelium.
Alternatively, adhesion molecules are not always necessary for niche colonization. Nonspecific adherence to epithelial surfaces via capsular or other bacterial surface proteins can occur, or bacteria can simply remain within the mucus layer. 88 This view is supported by the fact that adherence of M haemolytica to nasal mucus can be increased by enzymatic degradation of the protein and carbohydrate components of mucus. 11 M haemolytica–derived neuraminidase has been shown to exert a pathogenic role by degrading mucus components and thus enhancing adherence of M haemolytica to epithelium. 115 Retzer et al 102 proposed that outer membrane iron-binding proteins transferrin-binding protein A and B (Tbp A and Tbp B) could serve as adhesin molecules because their homologue in Neisseria meningitidis is expressed on the bacterial surface. 102 Furthermore, based on evaluation of the M haemolytica genome sequence and in comparison with the sequences from N meningitidis and Bordetella sp filamentous hemagglutinin, Highlander 52 proposed that a filamentous hemagglutinin homologue of high molecular weight (340 Kd) in M haemolytica is an adhesin molecule. In addition, several other gene sequences of putative adhesins were proposed after analysis of the M haemolytica S1 genome sequence; however, their role as adhesins has not yet been confirmed. 47,52
Lipopolysaccharide
LPS plays a critical role in the pathogenesis of SF. Current evidence indicates that LPS contributes to the pulmonary lesions through a variety of complex mechanisms, including the stimulation of leukocytes to produce proinflammatory cytokines, the activation of complement and coagulation cascade, and direct cell cytolysis. 69,83,85 The LPS molecule consists of polysaccharide side chain (O antigen), lipid A, and inner and outer cores of oligosaccharides, with the size of the immunogenic side chain determining whether the LPS type has a rough or smooth material. 8,67 The lipid A moiety of LPS is responsible for eliciting endotoxic effects, such as pyrexia and hypotensive shock. Although O antigen of LPS is immunogenic and antibodies exhibit cross-reactivity among different serotypes, there is unfortunately no correlation between high antibody response to LPS and protection of the host from development of pneumonia. 23,24
LPS is a potent vasodilator; it stimulates pulmonary endothelial changes consistent with the vascular leakage. 86 In addition, when administered intravenously in sheep, LPS produces clinical signs associated with the hypotension. 4 The systemic effects of LPS are considerably important in acute pasteurellosis, which causes septicemia in lambs, resulting in high mortality. The pathological effects of LPS are mediated by binding with LPS-binding protein, and further interactions are most likely mediated by CD14. LPS forms high–molecular weight complexes with LKT, enhancing the pathogenic effect of each other. 69,76 Additionally, in vitro studies revealed that bovine alveolar macrophages simultaneously challenged with LKT and LPS produced more tumor necrosis factor α (TNFα) and interleukin 8 (IL-8), compared to cells challenged with each factor individually. 69 There is some similarity between the effects mediated by LKT and LPS. Because LPS and LKT can physically interact and it is difficult to obtain purified LPS alone without LKT, it is possible that results of some in vitro studies using LPS or LKT alone are influenced by the presence of the other molecule.
In pneumonic calf lungs, LPS can rapidly cross the alveolar wall and become localized within the cytoplasm of neutrophils, alveolar macrophages, endothelial cells, and pulmonary intravascular macrophages and on epithelial cell surfaces. 124 LPS can stimulate alveolar macrophages to produce proinflammatory cytokines, reactive nitrogen intermediates, reactive oxygen intermediates, and other mediators that can actively participate in the inflammatory process; these include IL-1β, IL-8, leukotriene 4, prostaglandin E2, and TNFα from bovine leukocytes. 55,69,83,129,131 Subsequently, these proinflammatory cytokines and chemotactic mediators initiate influx of neutrophils. 13,69 In vitro challenge of bovine neutrophils with both LPS and LKT resulted in degranulation, generation of superoxide and nitric oxide, and lysis. 80 Another in vitro study demonstrated increased expression of elastase, myeloperoxidase, nitric oxide, and alkaline phosphatase in neutrophils. 123 The in vitro effect of LPS on bovine alveolar macrophages demonstrated a dose-dependent increase in iNOS gene expression, and nitric oxide generated from LPS-stimulated bovine alveolar macrophages caused cytotoxic injury to pulmonary endothelial cells in a dose-dependent manner. 131 Similarly, inoculation of M haemolytica in calf lung lobes revealed iNOS gene expression in leukocytes and epithelial cells. 101
LPS can cause pulmonary damage directly through toxic effects on pulmonary endothelium and indirectly through neutrophil recruitment. 55,85,97 The toxicity of LPS can be enhanced by complexing it with the phospholipids in the pulmonary surfactant, which allow LPS to persist in the lung and initiate inflammation. 12,90 Other systemic effects induced by LPS include fever and production of acute-phase proteins by the liver.
Similar to LKT, LPS exhibits a dose-dependent effect on the bovine peripheral blood leukocytes. At low concentrations, LPS decreases the phagocytic ability of neutrophils, whereas at higher concentrations, the phagocytic activity increases. 25 Moderate concentration is mitogenic for mononuclear cells, whereas a high concentration has the opposite effects. 25 Emau et al 41 studied the molecular mechanisms underlying these effects using purified M haemolytica LPS infusion. The authors concluded that cyclic nucleotides mediated the action of LPS at the cellular level, as evidenced by an increase in plasma arachidonic acid, thromboxane B2, and prostaglandin E levels.
Capsule
There are 12 serotypes of M haemolytica (S1, S2, S5–S9, S12–S14, S16, S17) based on the differences in capsular polysaccharide antigen typing. 103 Log-phase bacteria exhibit good encapsulation, whereas stationary-phase bacteria exhibit poor encapsulation. 27 The chemical structures of capsular polysaccharides of M haemolytica S1, S2, and S7 have been determined 2 and are similar to those from other pathogenic bacteria. S1 capsular polysaccharide structure is similar to the widely distributed enterobacterial common antigen; S2 capsular polymer is identical with capsular polysaccharides of N meningitidis B and Escherichia coli K1; and S7 structure is similar to polysaccharides associated with N meningitidis group L and Haemophilus influenza type F. 2 –4
The capsule of M haemolytica S1 may be involved in colonization of lung by promoting bacterial adherence to respiratory epithelium. 89 Except for lung colonization, the role of capsule in the pathogenesis is not yet established. Chae et al, among others, 14,31,32 demonstrated that the presence of capsule reduces leukocyte phagocytosis as well as complement-mediated lysis of the bacterium, whereas others have found that the presence of anticapsular monoclonal antibodies promotes phagocytosis but not complement-mediated killing. 127 Czuprynski et al 31 found that M haemolytica capsular polysaccharide stimulated release of IL-1 from bovine blood monocytes but not from alveolar macrophages. Vaccination of cattle with live M haemolytica or capsular polysaccharide promotes production of anticapsular antibodies; however, a positive correlation between capsular antibody and protection has not been established. 26,121
Outer Membrane Proteins
M haemolytica possess multiple outer membrane proteins. 96 Several of these proteins are iron-regulated outer membrane proteins, such as Tbp 1 and Tbp 2, and are physiologically and pathologically relevant because they are in involved in iron acquisition. 93 Because M haemolytica does not produce siderophores, expression of these iron-regulated outer membrane proteins is the main mechanism of iron acquisition. 62,93 Furthermore, Iovane et al 57 demonstrated that outer membrane proteins are chemotactic agents for neutrophils and inhibit their phagocytic and subsequent bacterial-killing mechanisms, thus favoring bacterial pulmonary colonization.
The protective antigens of M haemolytica are not completely understood; however, data indicate that protective immunity against M haemolytica can be acquired through the production of neutralizing antibodies against outer membrane proteins and LKT. 22,108 One of the immunogenic outer membrane proteins that offer protection following vaccination is a 45-kDa S1 outer membrane lipoprotein designated PlpE. 94,95 This outer membrane protein was identified in all serotypes, but its structural and physiologic function is not known. The antibodies generated against PlpE offer cross protection against serotype 6 and promote phagocytosis and complement-mediated bacterial killing. 94
Another outer membrane protein, OmpA, is a highly conserved protein that is involved in binding to specific host cell receptors in the upper respiratory tract, thereby playing a role in adherence and colonization and in generating host specificity. 37,63 Other studies have found that outer membrane proteins stimulate the release of nitric oxide, induce the expression of iNOS in interferon γ–activated macrophages, increase actin polymerization, and modify oxidative burst in neutrophils. 57,84
Toxins and Extracellular Enzymes
Leukotoxin
M haemolytica LKT and LPS are well-characterized virulence factors with respect to their pathogenic role in the SF. 52,60 Since its initial discovery, LKT has been the subject of intense investigation relative to its role in the pathogenesis of SF. LKT is an exotoxin and a member of the RTX (Repeats in ToXin) family of toxins. These toxins are genetically related and share a common, highly conserved motif consisting of a series of glycine–aspartic acid nonapeptide repeats in the carboxy terminal third of the LKT protein molecule. This conserved motif is involved in calcium binding and plays a vital role in inducing leukocyte toxicity because of its ability to form tertiary conformation required for target host cell binding. 29 The conserved motif also contains a recognition site required for transport of LKT across biological membranes in bacteria by the tolC-dependent type I secretion system. 28,128 The other members of this family and their LKTs include Actinobacillus actinomycetemcomitans (LtxA), Actinobacillus pleuropneumoniae cytotoxins (ApxI, ApxII, ApxIII and ApxIV), E coli alpha hemolysin, Actinobacillus suis subs haemolyticus toxin (Aqx), Fusobacterium necrophorum LKT, Bibersteinia trehalosi, and Bordetella pertussis hemolysin. 66,91,105,106 Although these members belonging to the RTX family of toxins are genetically related owing to the similarity of mechanisms involved in LKT activation, secretion, and partially shared amino acid sequences, they differ markedly in target cell specificity. 122
M haemolytica LKT is a 102- to 105-kDa protein produced by all serotypes during the logarithmic phase of bacterial growth in vitro. 107 The genetic organization of the LKT gene complex is composed of four genes, with genes lktC and lktA located upstream and genes lktB and lktD located downstream. 53 The structural gene product of lktA is composed of 953 amino acids, which encodes the structural yet biologically inactive LKT–protoxin. Gene lktC encodes transacylase that posttranslationally modifies the inactive LKT–protoxin by fatty acid acylation, thereby converting it into a biologically active LKT (LKT-A). During acylation, the fatty acid groups are added to the lysine residues located on LKT-A, which is a critical step in removing charge and increasing the hydrophobicity of LKT-A and which allows LKT-A to insert into host cells and form transmembrane pores. Although acylation is not required for LKT-A binding to the target cells, it is required for increasing intracellular calcium ion concentration, generation of reactive oxygen species, and production of IL-8 and for causing cytolysis. 119 Gene products of lktB and lktD are required for transport of LKT-A from the bacterial cytoplasm into the outer environment (Fig. 3).
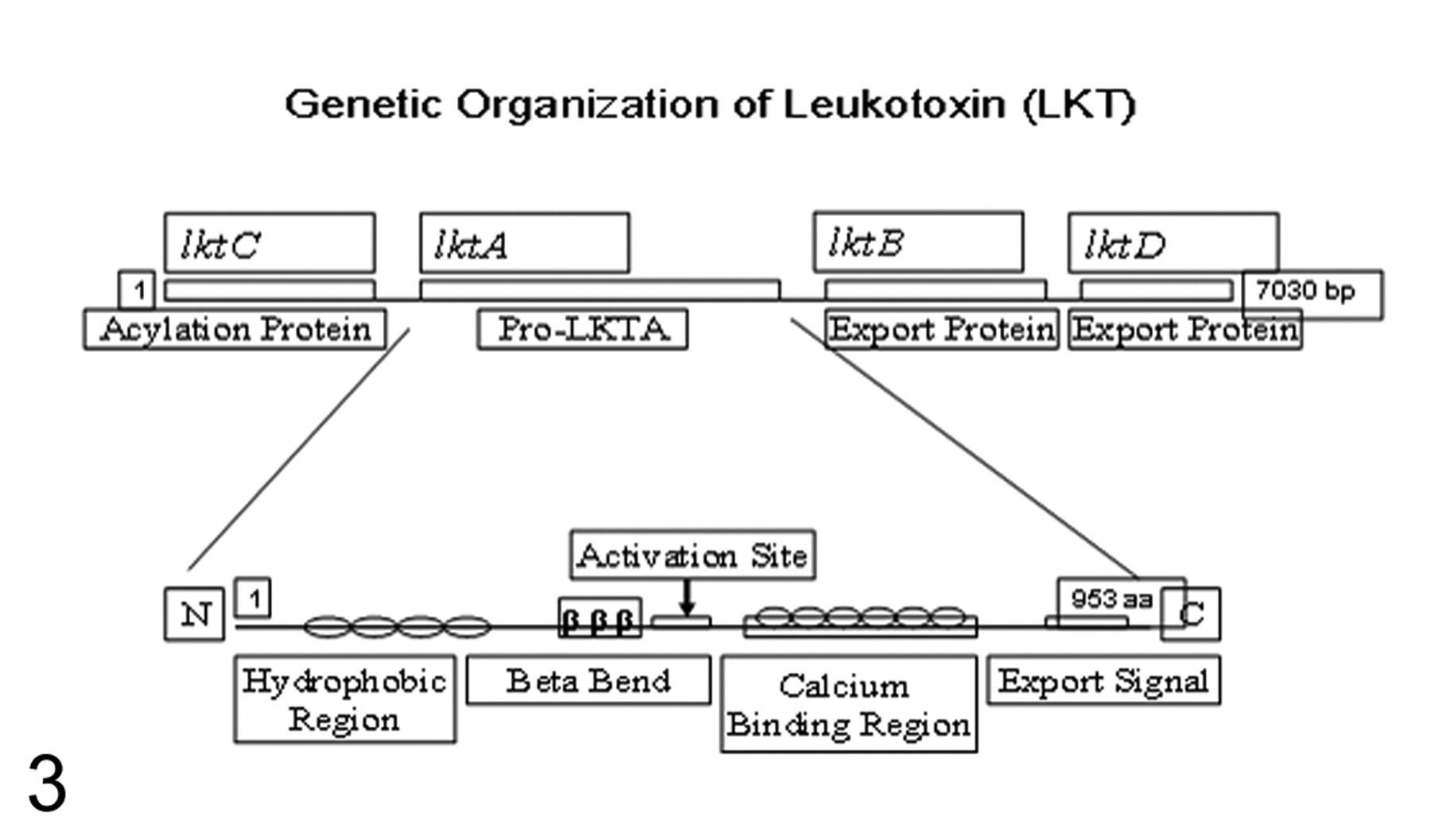
LKT-A contains domains that are responsible for receptor binding, pore formation, and calcium binding. 29,116 The N-terminal region is putatively involved in the receptor binding, whereas the adjacent residues of LKT-A consist of a series of hydrophobic residues implicated in spanning the host cell membrane and, possibly, pore formation. 91 The carboxy terminal domain of LKT-A contains glycine and aspartate-rich repeats, and neutralizing epitopes are present within a 229–amino acid region at the C-terminal end of LKT-A. 53
LKT follows a species-specific dose-dependent activation–inhibition paradox on bovine leukocytes. 91 At low concentrations, LKT can activate neutrophils and macrophages to stimulate respiratory burst and degranulation, proinflammatory cytokine (TNFα, IL-1, and IL-8) release from neutrophils and alveolar macrophages, and histamine release from mast cells, as well as reduce mitogen-mediated lymphoid proliferation. 53,80 At high concentrations, LKT stimulates bovine leukocytes to undergo apoptosis by engaging extrinsic and intrinsic mechanisms, whereas at highest concentrations, LKT causes transmembrane pore formation, cell swelling, and subsequent cell necrosis (oncotic cell death). 10,20,120 The transmembrane pores in the plasma membrane of activated macrophages and neutrophils and their subsequent oncotic cell necrosis cause leakage of products into the surrounding pulmonary parenchyma, contributing to the pulmonary damage—specifically, products of respiratory burst (ie, oxygen free radicals, superoxide anions, and hydrogen peroxide) and others, such as nitric oxide, lysosomal enzymes (including myeloperoxidase), and arachidonic acid metabolites (such as leukotriene B4 and 5-hydroxyeicosatetraenoic acid). 31,51,79,80,131 In addition, M haemolytica LPS can complex with LKT, enhancing LKT-induced cytotoxicity. 69,76
LKT-A is a key virulence factor that contributes to the pathogenesis of inflammation and pulmonary necrosis in SF, and it is specific for ruminant leukocytes. 54,117 In diseased lungs, LKT is associated with the cell membranes of degenerating inflammatory cells located in alveoli. 124 Although most strains of M haemolytica from cattle and sheep produce LKT, not all strains are equally pathogenic, because LKT from these strains do not exhibit similar leukotoxic activity and the amount of LKT produced is variable. 36,103,104 The importance of LKT in causing pulmonary necrosis was established by intratracheal inoculation of calves with a LKT-deficient mutant M haemolytica, which caused lower mortality and decreased lung lesions, as compared to animals challenged with the parent strain capable of producing LKT-A. 54,117
M haemolytica–induced pneumonia and other diseases are observed in only ruminants—including cattle, sheep, bighorn sheep, goats, bison, and exotic ruminants—because LKT-induced effects are specific for ruminant macrophages, lymphocytes, neutrophils, and platelets. 34,35 This species specificity stems from the selective interaction of LKT with the β2 integrin LFA-1 (lymphocyte function-associated antigen 1; CD11a/CD18) on target host cells. 35,40,48,59,71,77,78 Integrins are expressed on most cell types and are involved in cell–cell interactions and cell–extracellular matrix interactions, whereas subtype β2 integrins are expressed exclusively on leukocytes, including T lymphocytes, macrophages, neutrophils, monocytes, and dendritic cells. 6
Despite a consensus that LKT-induced specificity is due to β2 integrin binding, a complete agreement is lacking on the specific β2 integrin and subunits of β2 integrin required for receptor–ligand interaction. 34,70,71 Most scientific evidence supports LKT interaction with the CD18 subunit of LFA-1. 36 Furthermore, in vitro incubation of bovine leukocytes with proinflammatory cytokines (IL-1β and TNFα) results in increased expression of LFA-1 and a simultaneous increase in LKT binding, cytotoxicity, and apoptosis. 30,73,74 Similarly, in vivo and in vitro results indicate that bovine herpesvirus 1 infection increases susceptibility of the bovine leukocytes to LKT binding and cytotoxicity by increasing LFA-1 expression on neutrophils and peripheral blood mononuclear cells. 75 Recent studies using LKT-resistant mouse histiocytoma, human K562, and other cell lines demonstrated that these cell lines could be rendered susceptible to LKT effects by expressing cattle CD18 subunits on these cells.
However, other evidence suggests that LKT binds not only with the CD18 subunit of other β2 integrins but also with the CD11a subunit. 40,120 It appears that binding of LKT to CD18 is critical in eliciting the pathologic effects of LKT, and during this binding, LKT may interact with CD11a subunit. 34,36,40 It was recently suggested that LKT particularly binds to the integrin epidermal growth factor 3–like domain of bovine CD18. 39 Furthermore, interaction not mediated by β2 integrin may occur, not only with cells from nonruminant species, but with cells lacking β2 integrins, such as erythrocytes and platelets. 116 It is therefore possible that some effects are mediated by nonspecific receptor–ligand interactions.
In addition, in vitro studies have revealed that at lower doses, LKT can induce apoptosis, activate leukocytes, or inhibit lymphoid proliferation. 103 Those latter effects are partially abrogated by preincubation of lymphocytes with IL-1 and IL-2. 81,82 The mechanism underlying this response is unknown; however, the authors suggested failure of IL-2 production by lymphocytes or altered IL-2 receptor expression. 81,82 In addition, this effect raises interesting concerns on whether increased susceptibility to other bacteria or viruses in SF is due to the underlying lymphoid necrosis. However, lymphopenia is not a clinical feature of SF, and LKT is restricted to the lungs of affected animals with no systemic distribution, suggesting that LKT-induced inhibition of lymphoid proliferation may be only an in vitro phenomenon.
LKT-induced cytotoxicity of host leukocytes is characterized by the formation of transmembrane pores and, eventually, oncotic cell lysis. 19,20 LKT inserts its N-terminus into the host cell membrane to form a hydrophilic pore, but how these terminal transmembrane pores are formed is controversial. Studies have indicated that polymerization of LKT occurs on host cells, thereby forming transmembrane pores, whereas others have suggested that LKT-induced regulation of voltage gated channels and, thereafter, formation of transmembrane pores. 19,56,87 In addition, Atapattu and Czuprynski 9 recently demonstrated that LKT binds to the cell receptors and relocates to lipid rafts and clathrin-coated pits, resulting in LKT internalization and cytotoxicity. LKT-induced cytotoxicity has been shown to be associated with a caspase 1–dependent pathway, whereas LKT-associated apoptosis proceeds through a caspase 9–dependent pathway. 10
Irrespective of the basic process involved in the pore formation, these pores are 0.001 to 0.002 μm in diameter, allow rapid loss of intracellular K+ from the host cell cytoplasm, and internalization of Na+ molecules, causing colloid–osmotic imbalances. 19 Subsequently, water moves into the cell to correct the imbalance, resulting in rapid cell swelling. 20 Uncontrolled transmembrane calcium influx occurs and likely activates membrane phospholipases and proteases, or it may disrupt the cytoskeletal structure.
The outcome of these alterations is degradation of the cell membrane and subsequent cytolysis of leukocytes, as demonstrated by release of cytoplasmic lactate dehydrogenase. 20 Similarly, LKT can enhance ruminant platelet adhesion and activate and induce transmembrane pore formation in platelets by calcium-dependent mechanisms. 15,21,92 The transmembrane pores in the plasma membrane of activated macrophages and neutrophils and their subsequent oncotic necrosis may cause leakage of products of respiratory burst, lysosomal enzymes, and arachidonic acid metabolites into the pulmonary parenchyma. 33,79,80,131 These by-products cause damage to the pulmonary parenchyma, giving rise to fibrinous and necrotizing bronchopneumonia typical of SF (Figs. 1, 2). 79 LKT can also induce release of histamine from isolated bovine mast cells, causing increased vascular permeability and rapid influx of serum proteins and more LKT-susceptible neutrophils at the location of inflammation. 5 Because β2 integrins are present on lymphocytes, LKT-induced damage to these cells may allow bacteria to evade host adaptive immune responses.
The influence of calcium ions on the cytotoxic activity of M haemolytica LKT is critical because depletion of intracellular calcium ion by addition of a calcium chelator eliminated the cytolytic effect of low doses of the toxin. Likewise, addition of calcium to target bovine leukemia cell line (BL-3) cell cultures depleted of the divalent cations restored the cytolytic effect of the LKT. Addition of a calcium channel blocker resulted in dose-dependent protection of BL-3 from LKT-induced cytolysis. These results suggest that calcium positively influences the rapid initial phase of cell death resulting from exposure to the toxin. 55,130
Proteases
A number of proteases have been associated with M haemolytica. Although their physiological and pathological roles are not completely understood, a role has been suggested—namely, one of circumventing innate and adaptive immune response, thereby enhancing lung colonization and establishment of the disease. For instance, in the bovine respiratory tract, immunoglobulin A predominates in the upper respiratory tract, whereas immunoglobulin G (IgG) is the primary antibody in the lower respiratory tract. 99,126 Both IgG1 and IgG2 are believed to be important in defense against M haemolytica infection. Glycoprotease obtained from the culture supernatant of M haemolytica can selectively hydrolyze IgG1, thereby reducing opsonization-induced phagocytosis and bacterial killing; it can also selectively degrade host mucosal sialoglycoproteins. 1,72,94 Shewen et al 106 used this glycoprotease to demonstrate that vaccination of calves with the recombinant glycoprotease alone and in combination with M haemolytica culture supernatant vaccine enhances protection against experimental disease. The pathogenic function of this enzyme is unknown; however, adherence of bacteria to host epithelial cells and aggregation of platelets in the alveoli have been demonstrated, whereas glycoprotease activity can be potentiated by coincubation with the LKT. 92
All strains of M haemolytica produce the enzyme neuraminidase; however, its function is unknown. 44,115 Many respiratory pathogens, including Hemophilus influenzae, Streptococcus pneumoniae, and Pseudomonas aeruginosa, express neuraminidases that can cleave α2,3-linked sialic acids from glycoconjugates. Because mucosal surfaces are heavily sialylated, neuraminidases may modify epithelial cells by exposing potential bacterial receptors, enhance biofilm formation, and evade local innate immune response by cleaving salivary glycoproteins. 49,65,112 Therefore, it is possible that neuraminidase produced by M haemolytica may play a role in the initial colonization and evasion of antimicrobial killing and immune response.
Summary
The pathogenesis of SF is elaborate and complicated. M haemolytica S1 is a normal inhabitant of the upper respiratory tract of cattle. Under the influence of predisposing factors, which include stress induced by change in the environment (shipment, inclement weather, cold air) or by microbial agents (bovine herpesvirus 1, bovine parainfluenza virus 3, bovine respiratory syncytial virus, bovine viral diarrhea virus, Mycoplasma sp, and other bacterial infection), M haemolytica S1 rapidly replicates, resulting in inhalation of droplets containing bacteria in the lungs. In the alveoli, it initially interacts with the resident pulmonary alveolar macrophages that serve as primary line of host defense.
By the help of various virulence factors, M haemolytica can evade the innate and adaptive immune responses, colonize lungs, and establish the infection. The foremost of these virulence factors are LKT and LPS. In low subcytotoxic dose, LKT can activate macrophages through its interaction with the β2 integrin receptor, which is present not only on macrophages but also on other ruminant leukocytes, including neutrophils and lymphocytes. 80,84 This interaction of LKT and LPS with bovine leukocytes is followed by activation of leukocytes to undergo oxidative burst and release proinflammatory cytokines such as IL-1, IL-8, and TNFα, leading to accumulation of inflammatory cells in the lungs (Fig. 4). 69,83,90,126
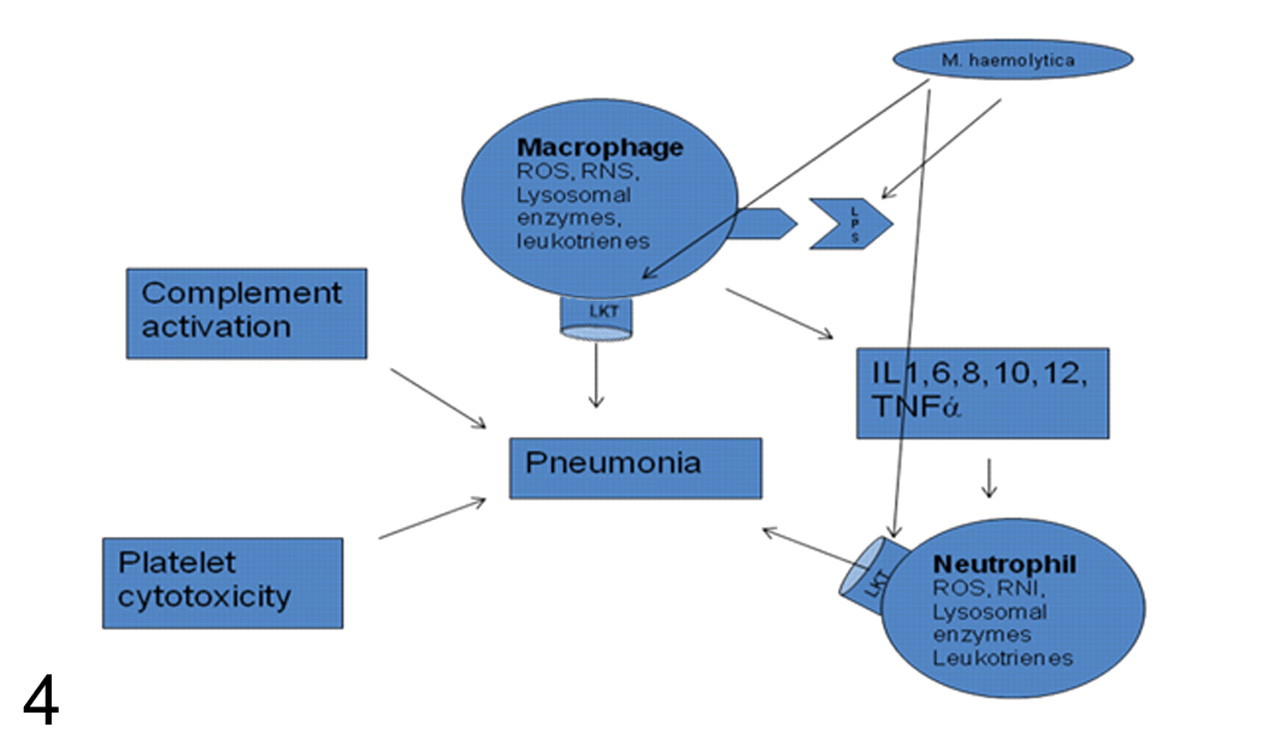
To overcome host defenses, M haemolytica has evolved a powerful mechanism of LKT-mediated cytotoxicity of the leukocytes involved in the innate and adaptive immune responses. 18 LKT forms transmembrane pores in bovine lymphocytes, neutrophils, macrophages, mast cells, and platelets, eventually causing oncotic cell cytotoxicity (Fig. 4). 16,18,20 These transmembrane pores and the cell cytolysis of activated leukocytes possibly cause leakage of oxygen radicals and other products, such as nitric oxide, lysosomal enzymes, and inflammatory mediators, into the surrounding pulmonary parenchyma, contributing to the pulmonary necrosis. 30,79,80 Moreover, increased expression of LKT receptor β2 integrin was observed on bovine neutrophils incubated in vitro with LKT, LPS, and cytokines such as IL-1β and TNFα, thereby increasing LKT binding to the leukocytes and, subsequently, cytotoxicity. 74,114 The biological effects of these inflammatory molecules and LKT are morphologically evident in the form of necrotizing bronchopneumonia and formation of neutrophil-derived oat cells typical of M haemolytica infection. 79,80 To circumvent host-adaptive defense mechanisms of antibody-mediated bacterial phagocytosis, M haemolytica has generated a unique method of cleaving IgG delivered to the site of inflammation by using its immunoglobulin proteases. 72 In addition, in vitro studies have revealed that LKT at sublethal doses is capable of inhibiting mitogen-induced lymphoid proliferation, potentially inhibiting adaptive immune responses. 81,82
Footnotes
Acknowledgement
We appreciate the comments by anonymous reviewers. We also acknowledge Dr W. E. Hoffmann for editing this article.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.