
Abstract
The mammalian immune system is remarkable in that it can respond to an essentially infinite number of foreign antigens. The ability to mount a long-lasting (adaptive) immune response against foreign antigen requires the participation of cells selected from an enormously diverse population of B and T cells. Because the B and T cell receptors expressed by these cells are generated at random, a significant percentage of B and T cells are invariably directed against self-antigen. Under normal circumstances, autoreactive B and T cells are eliminated, reprogrammed, or inactivated in the primary and secondary lymphoid organs. Despite these checks and balances, a small but significant number of people and animals still develop autoimmune disease. One such autoimmune disease—systemic lupus erythematosus—is characterized by the loss of B- and T-cell tolerance to self-antigens (principally nuclear), culminating in multisystemic inflammation. Multiple genetic defects, drug exposure, infectious agents, and environmental factors can contribute to the pathogenesis of the disease. Loss of B- and T-cell tolerance precipitates activation of plasmacytoid and myeloid dendritic cells; collectively, these cells cooperate to form a complex positive feedback loop, continually stimulated by the persistence of self-antigen. Novel treatment strategies now focus on specific inhibition of various aspects of the feedback loop. These specific inhibitors have the potential to be more effective and lack the side effects associated with generalized immunosuppression.
The mammalian immune system is a remarkable product of evolution. It has the capability to mount a long-lasting (adaptive) immune response to virtually any foreign antigen it encounters, which requires the presence of an enormously diverse population of T and B cells. During normal maturation, T- and B-cell receptors are generated through a series of genetic recombination events yielding lymphocytes with the required receptor diversity. Because this is a somewhat random process, a significant percentage of the cells generated invariably have T and B receptors directed against self-antigen. Under normal circumstances, those T and B cells with high affinity for self-antigen are eliminated, reprogrammed, or inactivated in the primary and secondary lymphoid organs. 42 However, these processes do not delete T and B cells with lower affinity for self-antigen. In fact, between 25 and 40% of circulating T cells recognize self-antigen and have the potential to induce autoimmunity. 13 Despite these high numbers of autoreactive lymphocytes present in circulation, about 3 to 5% of the human population actually develops an autoimmune disease. 7
One such autoimmune disease—systemic lupus erythematosus (SLE)—is recognized in a variety of species, including mice, humans, and various domestic animals. SLE is characterized by the loss of B- and T-cell tolerance to one or more self-antigens, resulting in polysystemic inflammation. SLE is known as the “great imposter,” and the spectrum of disease symptoms present in each patient depends on underlying genetic defects and the anatomic location in which the humoral and cellular responses against self-antigen are manifested. Patients experience a confusing array of clinical symptoms, including any combination of renal disease (glomerulonephritis, interstitial nephritis, vasculitis, and proteinuria), swollen joints, skin rash, hematologic disorders, respiratory, and neurologic dysfunction (reviewed in Rahman and Isenberg 92 ). Further revealing the complexity of this disease, numerous susceptibility genes have been defined in humans and mice. 36,69,93,116 Given the large number of susceptibility genes potentially involved in the pathogenesis of SLE, it is possible that a different combination of these genes uniquely contribute to the disease pathogenesis in each patient. Such variation may partially account for the different symptoms and disease severity observed across the population. However, this complexity may allow for therapeutic intervention at many levels. Here we discuss how mouse models have contributed to our understanding of the pathogenesis of SLE, and we describe some therapies on the horizon.
Common Mouse Models of SLE
Mouse models have significantly contributed to our understanding of the pathogenesis of SLE. Unlike most other mouse models of autoimmunity and inflammation, SLE in mice closely resembles human SLE, including autoantibody production and renal disease. Congenic strains have allowed researchers to study the complicated genetics of SLE, both polygenic and at the single-gene level. There are many mouse models of SLE, and choosing the most appropriate for a question of interest requires an understanding of the clinical, pathological, and genetic features of each model. There are two main categories of mouse models of SLE: spontaneous (and their congenic counterparts) and induced (which includes gene targeting). Here, we present and contrast the main features of the best-characterized spontaneous and induced models.
The most commonly used mouse strains that develop spontaneous disease include the F1 cross between New Zealand Black and New Zealand White (NZB/W) mice, MRL/lpr mice, and BXSB/Yaa mice. Multiple genes contribute to the development of SLE in NZB/W mice, including major histocompatibility complex (MHC) and several non-MHC genes. 64 In addition to having a background of genetic susceptibility, MRL/lpr and BXSB/Yaa strains each have single genetic mutations that accelerate SLE. The MRL/lpr strain contains the loss of function lpr mutation in the gene for Fas. Without Fas–Fas ligand interactions, apoptosis of B and T lymphocytes does not occur, resulting in massive lymphoproliferation that contributes to the observed acceleration of disease. 125 In general, in human and most mouse models of SLE, females are more susceptible to disease. However, the opposite is the case for BXSB/Yaa mice, in which SLE develops earlier and is more severe in males than females. Although the BXSB genetic background confers SLE susceptibility to both sexes, males in this strain contain the Yaa mutation (Y chromosome–linked autoimmunity accelerator). This mutant locus consists of a duplication of at least 17 genes from the X chromosome, including toll-like receptor 7 (TLR7; discussed below). The location of this mutation on the Y chromosome explains the enhanced male susceptibility to disease. 50,91,114 Thus, as with human SLE, no single gene in the spontaneous models is responsible for mouse SLE.
The common immunological and clinical manifestations of SLE in these 3 strains include hyperactive B and T cells (their presence and interactions with each other are required for disease), high titers of several autoantibodies directed against nuclear antigens, defective clearance of immune complexes, and fatal immune glomerulonephritis. BXSB/Yaa male mice tend to have the most severe nephritis, followed by MRL/lpr mice and NZB/W female mice. In the former 2 strains, about 90% of mice are dead by 8 to 9 months versus 50% of NZB/W females. In addition to exhibiting hyperactive or abnormal B and T cells, BXSB/Yaa mice exhibit monocytosis and lymphoid hyperplasia; however, the lymphoid hyperplasia is not as massive as that seen in MRL/lpr. In fact, lymphoid hyperplasia is not a feature of human lupus and can confound histopathology. Despite the lymphoid hyperplasia, there are several unique features of the MRL/lpr strain. Compared to other strains, it has the highest titers and largest array of autoantibody specificities, and it is the only strain to develop antibodies against the nuclear components Smith (Sm) and ribonucleoprotein. MRL/lpr mice are also unique in that a proportion of mice develop skin rash, inflammatory arthritis, and immunoglobulin (Ig) M rheumatoid factors. Interestingly, as discussed below, arthritis and rheumatoid factors, as well as the development of anti-Sm antigen and anti-small nuclear ribonucleoprotein antibodies, are also present in pristane-induced lupus (Table 1 ).
Characteristics of Common Spontaneous Lupus Mouse Strains and Pristane-Induced Lupus in Mice a
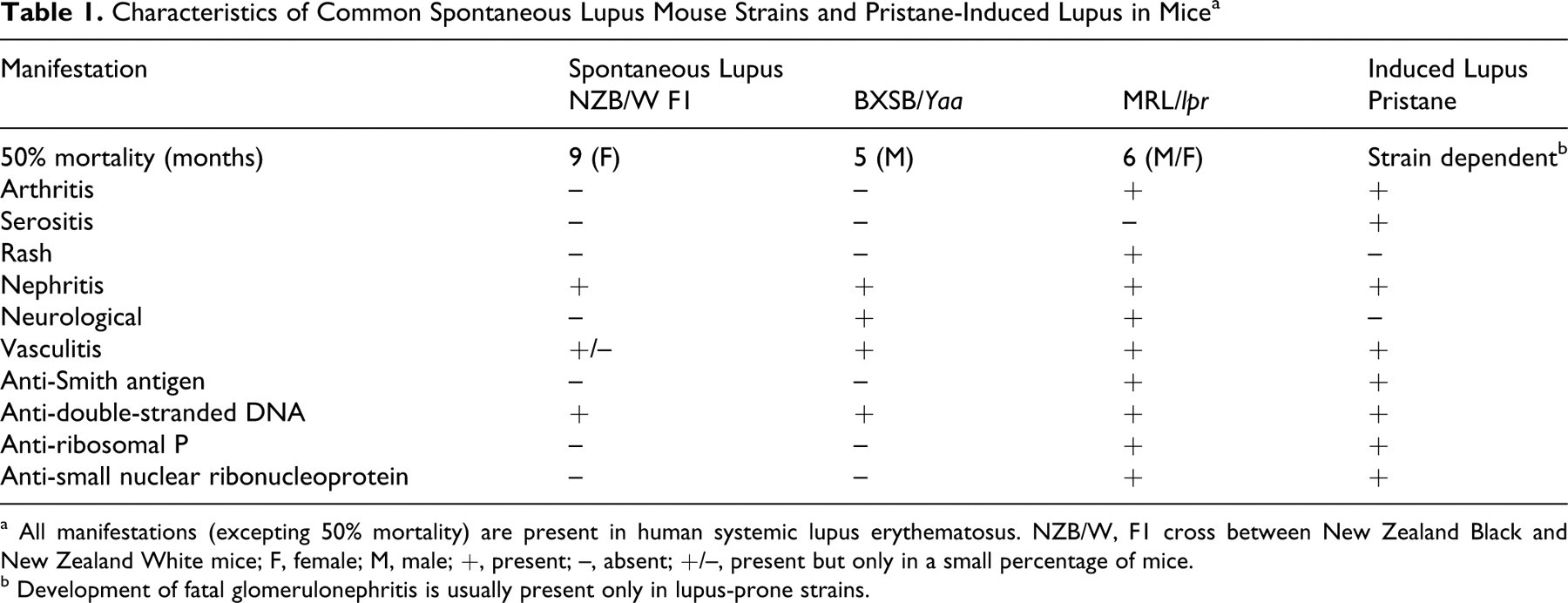
a All manifestations (excepting 50% mortality) are present in human systemic lupus erythematosus. NZB/W, F1 cross between New Zealand Black and New Zealand White mice; F, female; M, male; +, present; –, absent; +/–, present but only in a small percentage of mice.
b Development of fatal glomerulonephritis is usually present only in lupus-prone strains.
Induction of lupus in normal mouse strains can be accomplished by a number of methods, such as genetic manipulation of single genes (either overexpression or deletion), injection of autoimmune sera or lymphocytes from SLE-prone mice, vaccination with apoptotic debris-laden dendritic cells, immunization with prototypical lupus antigens such as DNA- and RNA-protein complexes or other antigens known to induce lupus, or injection of pristane (a hydrocarbon oil). 94,101 Strains that develop spontaneous disease and induced lupus models can be used to test pharmacological agents in vivo. However, to test the effects of single genes within the genetic background of a spontaneous strain requires significant time and resources to backcross transgenic or knockout mice onto the susceptible strain. Induction of lupus with a single injection of pristane results in a disease with most of the features of human lupus (Table 1). Because pristane can induce lupus in almost any strain, 102 it can save significant time in determining the effects of single genes on lupus development in transgenics and knockouts on conventional backgrounds. In addition, pristane injection in spontaneous models can be used to expand the repertoire of antibody responses and exacerbate nephritis. 128
Although induced models of SLE have been useful, much of our knowledge of the pathogenesis of SLE has been derived from careful study of the spontaneous disease models. Accordingly, the following sections emphasize the contributions of the spontaneous models.
Autoreactive B and T Cells Play a Role in the Pathogenesis of SLE
SLE is thought to be caused by a loss of B- and T-cell tolerance. Autoreactive B cells were first implicated in the pathogenesis of SLE in the 1950s when anti-DNA antibodies were recognized as a central component of the disease, and recent experiments have further proven the point. Additionally, homozygous B-cell deficient progeny of SLE-prone MRL/lpr mice bred to mice with the Jh (B-cell deficient) mutation are protected from developing disease. 111 Similarly, SLE-prone (New Zealand White mice × BXSB)F1 mice depleted of B cells are protected from developing disease. 18 T cells also appear to participate in the pathogenesis of SLE. Spontaneous T-cell activation is a characteristic of disease: 19 Tissues from mice 19,130 and humans 34 with SLE are infiltrated with CD4+ T cells, and these infiltrates are composed of a limited number of T-cell clones. 130 Also, MRL/lpr mice that are genetically deficient in CD4+ T cells or MHC class II molecules (and therefore deficient in the production of CD4+ T cells) develop lymphadenopathy, produce significantly reduced amounts of autoantibody, and do not develop renal disease. 56,62 These observations suggest that B and T cells are involved in the pathogenesis of SLE, but they do not explain the contribution of each to disease progression. The mechanisms underlying the loss of self-tolerance and the interaction between autoreactive B and T cells in individuals afflicted with SLE have been topics of intensive study for the past two decades and are discussed below.
The Adaptive Immune Response in SLE Patients Is Different From Non-SLE Individuals
There are important differences in the adaptive immune response of SLE patients and non-SLE individuals. In the latter, B- and T-cell-mediated immune responses are directed against foreign antigen, as tempered by the simultaneous induction of a counterregulatory response (ie, regulatory T cells), and the responses abate when the foreign antigen is cleared (reviewed in Brusko et al 17 and Torgerson 117 ). In contrast, the B- and T-cell-mediated immune responses in SLE focus on self-antigen; the regulatory T-cell response may be reduced or lacking; 65 and the autoantigens to which the patient is responding are never cleared. In fact, there is ample evidence that the SLE immune response is essentially a positive feedback loop in which participating immune cells are persistently activated either directly or indirectly by the presence of autoantibody–antigen complexes 22 (Fig. 1 ). Once initiated, the autoimmune response can spread beyond the inciting autoantigen to other autoantigens (ie, epitope spreading), thus exacerbating disease. 29 Thus, SLE patients are characterized by a loss of B- and T-cell tolerance to self-antigen, leading to autoantibody production and multisystemic inflammation. The reason for this loss in self-tolerance remained a mystery for many years, but research during the past two decades has revealed a number of mechanisms that individually or in combination contribute to the onset of SLE.
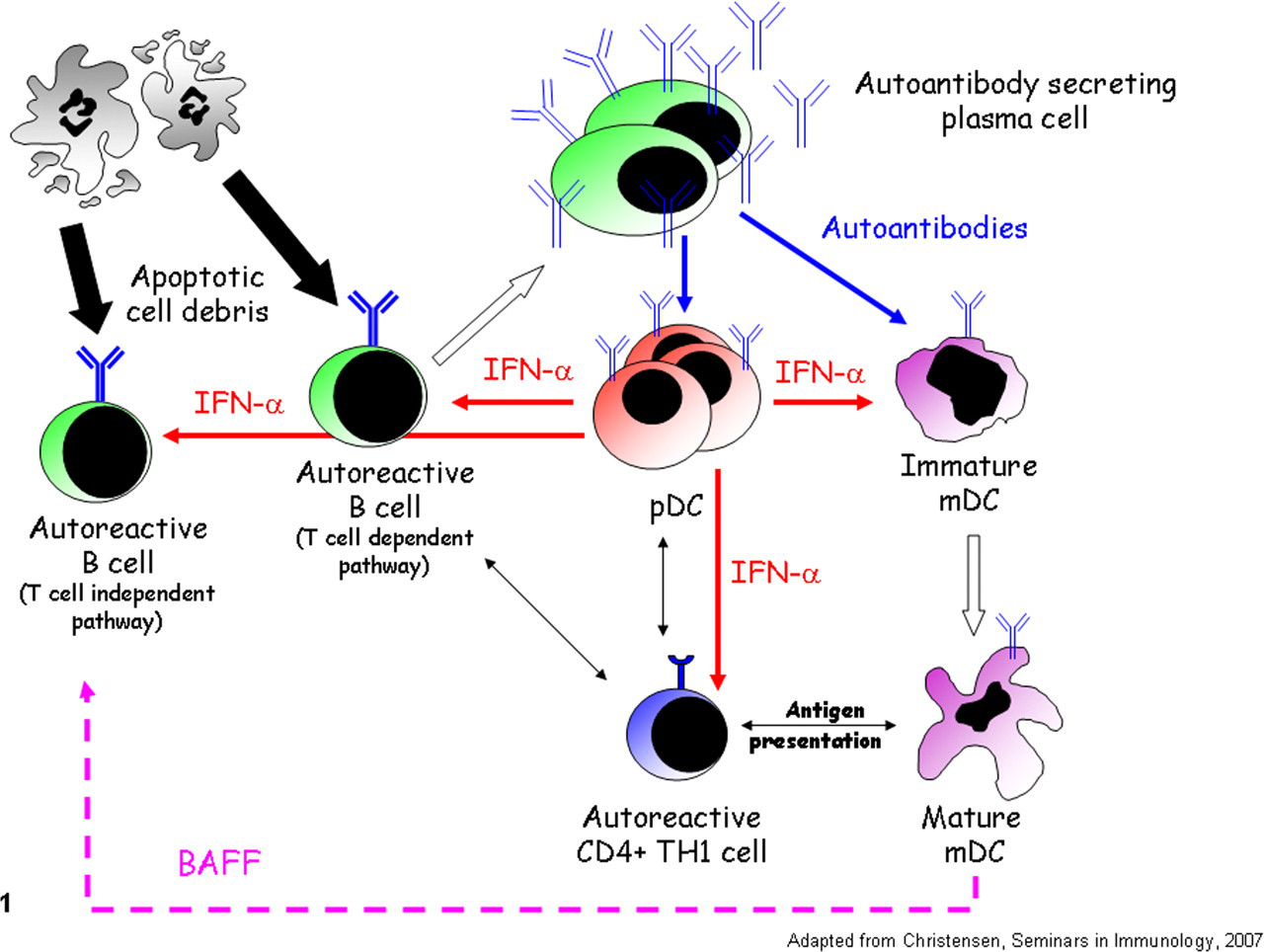
The autoimmune response in systemic lupus erythematosus patients is amplified by a positive feedback loop: During early disease, autoreactive naïve B cells bind autoantigen (apoptotic debris) and present the antigen to autoreactive T cells (T cell–dependent pathway). Following mutual activation, copious autoantibody is produced that binds to Fc receptors on plasmacytoid dendritic cells (pDCs) and myeloid dendritic cells (mDCs), rendering them responsive to autoantigen. After autoantigen exposure, pDCs produce interferon α (INF-α), which accelerates B-cell maturation and antibody production, autoreactive T-cell activation, and the maturation of mDCs. Likewise, autoantigen exposure activates mDCs to produce copious amounts of the cytokine BAFF. Both pDCs and mDCs present autoantigen to autoreactive T cells, further amplifying the inflammatory response. During disease progression, autoantibody production becomes independent of T cells because autoreactive B cells are directly activated by combined stimulation through toll-like receptor 7/9 and the BAFF and IFN-α receptors.
Autoimmune Response in SLE Patients Focuses on a Limited Number of Autoantigens
Despite the range of clinical symptoms associated with SLE, in both mice and humans, autoantibodies are primarily directed against a relatively small number of autoantigens, mainly of nuclear origin (Table 2 ; reviewed in Rahman and Isenberg 92 ). Antibodies are not only formed against DNA, ribonucleoproteins, and phospholipids but directed against the resultant immune complexes. Although the reason why human SLE patients develop autoantibodies against nuclear antigens remains to be clarified, infection with Epstein-Barr virus has been identified as a risk factor. 49 One recent hypothesis is that, following infection, SLE patients initially develop antibodies directed against Epstein-Barr virus nuclear antigen–1 that cross-react with the ribonucleoproteins Sm and Ro, thus forming antibody–autoantigen immune complexes. The ensuing immune response against these antibody–autoantigen complexes promotes epitope spreading, resulting in the production of antibodies against a host of new nuclear antigens, 48 including nucleosomes. 84 Interestingly, nucleosomes from apoptotic cells, not living cells, are able to activate antigen-presenting cells (APCs) 120 and are potent autoantigens for both B and T cells. 16 The ability of apoptotic cell-derived nucleosomes to induce APC activation critically depends on the presence of the proinflammatory mediator HMGB1 (high-mobility group box protein 1), which is not found in nucleosomes derived from living cells. Nucleosome–HMGB1 complexes are present in the circulation of SLE patients, suggesting that such patients have a high endogenous rate of apoptosis. 120 Thus, exposure to an infectious agent and the development of antiviral antibodies that cross-react with nuclear antigens in apoptotic cells is one mechanism by which SLE patients may initially develop autoantibodies. Additional predisposing factors have been hypothesized to contribute to autoimmunity, as discussed below.
Autoantibodies in Human Systemic Lupus Erythematosus a

a Adapted from Rahman and Isenberg. 92
b Ribonucleoproteins.
SLE Patients Have Defective B- and T-Cell Tolerance to Nuclear Antigens
Why do SLE-prone mice and human SLE patients develop autoimmunity to nuclear antigens? One reason is that there may be a central B-cell tolerance defect. For example, in normal mice 35,100 and healthy humans, 30 immature B cells with specificity for nuclear antigens undergo anergy or receptor editing, thus inducing tolerance. In contrast, MRL/lpr mice fail to delete or inactivate autoreactive B cells with specificity for DNA/chromatin 98 and RNA, suggesting a defect in central tolerance induction.
Peripheral B-cell tolerance may also be defective in SLE patients. Regulatory T cells play an important role in peripheral tolerance, and depletion of regulatory T cells leads to exuberant autoantibody production. 83 In contrast, reconstitution of the regulatory T-cell population can suppress autoantibody production in vitro and in vivo. 67,106,110 One mechanism by which regulatory T cells inhibit autoreactive B-cell function in both mouse and human is by inducing apoptosis through a direct cell–cell contact mechanism. 51
Human SLE patients 113 and SLE-prone NZB/W mice 57 may have a deficiency in the expression of the inhibitory receptor Fc gamma receptor IIB (FcγRIIB) on the surface of their B cells. The strength of FcγRIIB signaling is critical for the induction of immune tolerance or autoimmunity. 113 Even normal C57BL/6 mice made deficient in FcγRIIB develop antinuclear antibodies, glomerulonephritis, and SLE-like disease. 12 Conversely, retroviral-induced overexpression of FcγRIIB in B cells of BXSB mice restores tolerance and improved survival. 81
In addition to defective B-cell control, SLE patients may have functional defects in T-cell tolerance that predispose to autoimmunity. For example, patients newly diagnosed with SLE have decreased numbers of circulating CD4+CD25+FoxP3+ regulatory T cells compared to controls, and these cells are deficient in their ability to suppress proliferation of autologous CD4+ and CD8+ T cells. 76 Thus, in SLE patients and in SLE-prone mice, the loss of tolerance to self-antigen is multifaceted. However, a number of additional genetic and functional abnormalities may predispose SLE patients to disease, as described below.
SLE Patients Have Hypomethylated DNA and an Increased Rate of Apoptosis
Epigenetic regulation is one mechanism by which genes are inactivated or activated at the level of DNA by altering the methylation status of specific cytosine residues, typically in the promoter region. 103 Methylation prevents gene transcription, whereas hypomethylation may activate gene transcription. 85 Interestingly, DNA extracted from T cells of SLE patients is hypomethylated, compared to T-cell DNA from non-SLE individuals. 90,96 Additionally, treatment with drugs that inhibit DNA methyltransferase activity, such as procainamide, or other drugs or agents that induce DNA hypomethylation, such as hydralazine, can promote anti-DNA antibody production in non-SLE individuals and lead to symptomatic flares in SLE patients. 121 Interestingly, in vitro treatment of normal human and mouse CD4+ T cells with the DNA methyltransferase inhibitor 5-aza-deoxycytidine promotes DNA hypomethylation, increases perforin expression, 74 and promotes cellular activation and loss of tolerance to self-MHC class II molecules. 61 The resultant MHC class II–specific CD4+ effector T cells attack and kill APCs in vitro, including macrophages. 61 Because SLE patient DNA is hypomethylated, a similar anti–MHC class II response could occur in vivo, resulting in apoptosis of MHC class II APCs. In support of this notion, lupus-prone (SWR × New Zealand Black)F1 mice have an increase in the number of apoptotic splenic dendritic cells. 60 Furthermore, human SLE patients have accelerated apoptosis of monocyte–macrophages, and this apoptosis is in part mediated by an autoreactive subset of T cells. 28 Thus, DNA hypomethylation in T lymphocytes from SLE patients promotes activation of autoreactive T cells, an increase in the rate of apoptosis, accumulation of apoptotic debris, and a simultaneous decrease in the population of cells normally charged with the clearance of the apoptotic debris (reviewed in Richardson 95 ). As discussed below, abnormal apoptosis may be a significant initiating factor in SLE.
SLE Patients Have Defective Clearance of Apoptotic Debris
In addition to loss of phagocytic cells, other mechanisms may lead to defective clearance of apoptotic debris. For example, a predisposing factor for SLE in mice and humans is genetic deficiency of complement components C1q, C4, and/or C2. Such deficiencies in the classical complement pathway may prevent C3 conversion and result in impaired clearance of immune complexes and apoptotic debris (reviewed in Truedsson et al 119 ). Additionally, genetic deficiencies in either DNase I 86 or surfactant protein D 89 may impede clearance of naked DNA and/or nucleosome immune complexes. Collectively, an enhanced rate of apoptosis, a decreased clearance of apoptotic debris owing to reduced numbers of phagocytic cells, and an impaired function of complement and other clearance mechanisms lead to persistent exposure to nuclear antigens. This persistence of nuclear antigens may predispose to the loss of self-tolerance.
Initial Autoantibody Production Depends on the Interaction of Autoreactive B and T Cells
Antibody production can broadly be divided into two categories: T cell–dependent production, which involves the participation of CD4+ T cells, and T cell–independent production, which does not. Initially, the production of autoantibodies in SLE appears to be T cell dependent. Autoreactive B cells bind, internalize, and present autoantigen in the context of MHC class II molecules to autoreactive CD4+ T cells (specific for the same antigen), which recognize the MHC–antigen complex via their T-cell receptor. 19,80 Simultaneously, the autoreactive B cells upregulate plasma membrane expression of the B7 family members CD80 and CD86, which bind CD28 onto the surface of the autoreactive T cell (Fig. 2A). The combined signals through the T-cell receptor and CD28 induce T-cell activation and cytokine expression, which in turn promote IgM production by the B cell (Fig. 2B). 38 During continued antigenic stimulation, autoreactive T cells subsequently upregulate the costimulatory molecules CD40L and ICOS, which interact with CD40 and B7RP.1, respectively, on B cells (Fig. 2C) and promote further B-cell activation and antibody isotype class switching to IgG and accelerate antibody production. The notion that SLE initially depends on T cell–mediated autoantibody production is supported by the fact that prophylactic blockade of the CD28/CD80, CD28/CD86, CD40L/CD40, or ICOS/B7RP-1 pathways can decrease or eliminate autoantibody production and inhibit disease onset and/or progression. 26,31,37,53,124 Although the antigen-presenting function of B cells appears important to the pathogenesis of SLE, other B-cell functions, such as cytokine production, may contribute to disease progression in SLE and other autoimmune diseases. 15
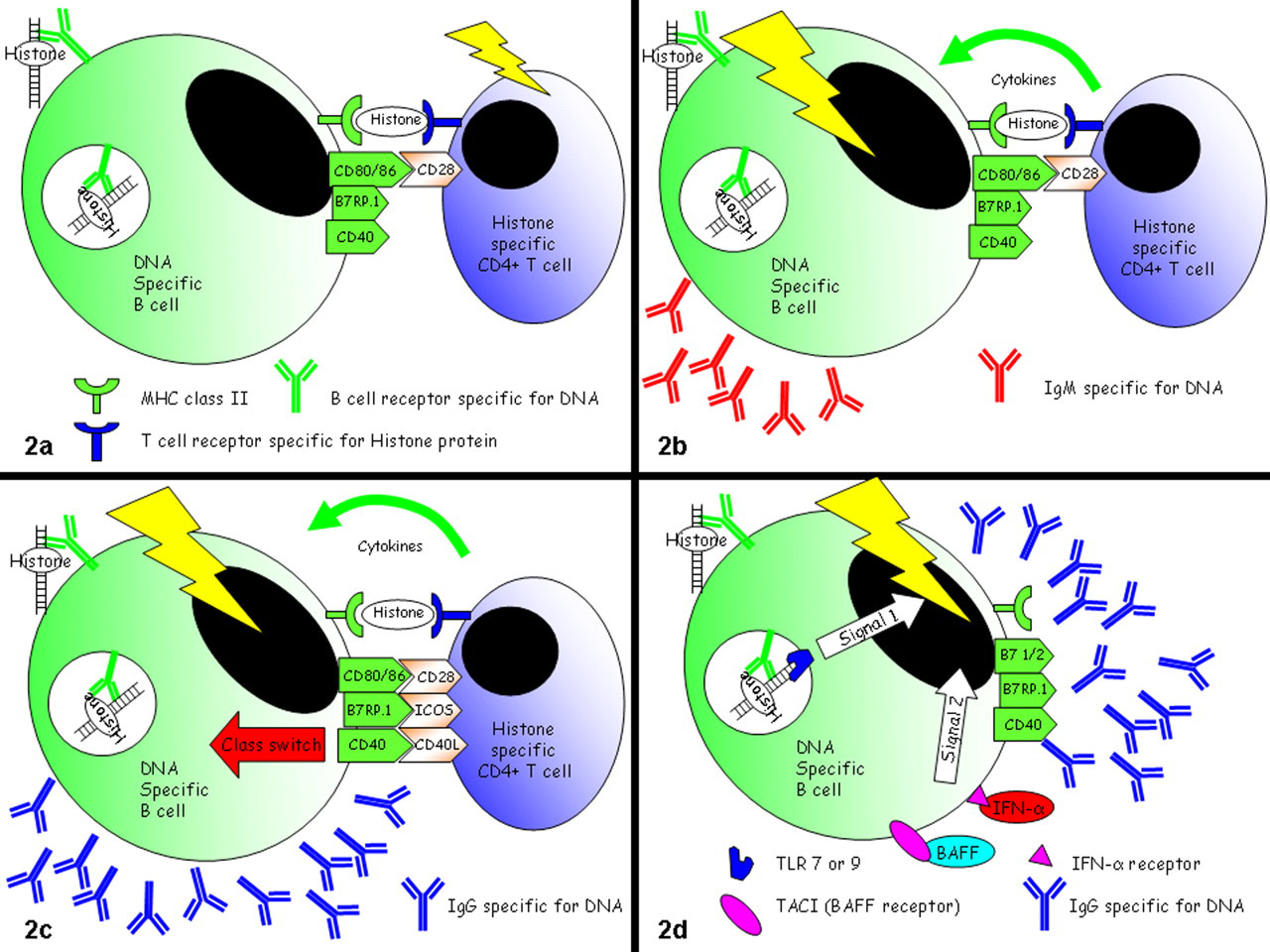
Activation of autoreactive B and T cells: Autoreactive B cells initially bind, internalize, and present autoantigen in the context of major histocompatibility complex (MHC) class II molecules to autoreactive CD4+ T cells (specific for the same antigen) that recognize the MHC–antigen complex via their T-cell receptor (T cell–dependent pathway). A, simultaneously, the autoreactive B cells upregulate plasma membrane expression of the B7 family members CD80 and CD86, which bind CD28 on the surface of the autoreactive T cell. B, the combined signals through the T-cell receptor and CD28 induce T-cell activation and cytokine expression, which in turn promote immunoglobulin M (IgM) production by the B cell. C, during continued antigenic stimulation, autoreactive T cells subsequently upregulate the costimulatory molecules CD40L and ICOS, which interact with CD40 and B7RP-1, respectively, on B cells and promote further B-cell activation and antibody isotype class switching to IgG and accelerate antibody production. D, as disease progresses, autoreactive B cells can be directly activated (T cell–independent pathway) by a combination of signals including toll-like receptor 7/9 recognition of nuclear antigen–antibody complexes, BAFF binding to the BAFF receptor TACI, and interferon α (IFN-α) binding to its receptor.
In SLE Patients, Apoptotic Debris Aberrantly Activates the Innate Immune System via Toll-like Receptors
Because apoptosis is a normal physiologic process, it is unclear why individuals who are normally tolerant develop autoimmune responses to their own nuclear components. One hypothesis is that in SLE patients, apoptotic products are processed in a different way to cause them to be immunogenic. Alternatively, if the apoptotic debris is phagocytosed and processed in an inflammatory environment, subsequent presentation by locally activated APCs to autoreactive T cells may precipitate the breakdown in tolerance. Finally, another possibility is that immune cells have specific receptors for DNA and RNA that are aberrantly activated in SLE patients. Interestingly, the latter hypothesis appears to be true. In an important article, Rifkin and colleagues demonstrated that certain mouse B cells (which express anti-mouse IgG2a receptors) are activated in vitro by IgG2a immune complexes containing nucleosomes but not by IgG2a isotype control antibodies, suggesting that the nucleic acid component of the complex is activating the cell. 97 Researchers subsequently demonstrated that these chromatin–IgG2a complexes activate B cells by dual engagement of the B-cell receptor and certain TLRs. 71 Two of these receptors are activated by nuclear antigens: TLR7 is activated by RNA and TLR9 is activated by DNA. It is now established that autoreactive B cells directed against RNA and DNA express both B-cell receptors for those molecules and the TLRs that are activated by RNA and DNA components (Fig. 2D). 21 –23 Thus, in SLE patients, DNA and RNA released from apoptotic cells are not only autoantigens but, actually, autoadjuvants. 68
Multiple lines of evidence indicate that activation of B cells through TLR7 and TLR9 is essential to the pathogenesis of SLE. First, TLR7-deficient MRL/lpr mice are protected from developing SLE. 23 Likewise, TLR9 deficiency also has a profound impact on disease and can either suppress autoantibody production 33 or exacerbate disease 22 depending upon themouse strain studied (56R+FcRIIB−/− mice vs MRL/lpr mice, respectively). Also, MRL/lpr mice deficient in MyD88, an intracellular adapter protein involved in TLR signaling, are protected from developing nephritis. 99 The importance of TLRs to the pathogenesis of SLE was further demonstrated in the BXSB mouse strain in which male mice develop severe lupuslike disease. As previously mentioned, these mice bear the Y chromosome–linked autoimmune acceleration (Yaa) mutant locus consisting of a duplication of at least 17 genes from the X chromosome, including TLR7, and the location of this mutation on the Y chromosome explains the enhanced male susceptibility to disease. 27 Reduction of TLR7 gene dosage abolishes the Yaa (lupus) phenotype, whereas an increase in gene dosage exacerbates disease. 27 In addition, with a pharmacological approach, treatment of lupus-prone mice with a TLR7/TLR9 inhibitor reduces autoantibody production and ameliorates symptoms of disease. 9 Finally, Komatsuda recently demonstrated that TLR7, TLR9, and interferon alpha (IFNα) mRNA are increased in the peripheral blood mononuclear cells of human SLE patients. 63 Thus, SLE patients not only have excessive apoptotic debris but may also be rendered hyperresponsive to their own apoptotic products by overexpression of TLR7 and TLR9.
Plasmacytoid Dendritic Cell–Derived IFNα Contributes to the Pathogenesis of SLE
Constitutive high-serum IFNα concentrations are a heritable risk factor for SLE. 88 In addition, studies show that IFNα administration to patients with malignancies or viral disease induces the expression of anti-DNA antibodies. 105 Plasmacytoid dendritic cells (pDCs) are a key source of IFNα; ligation of TLR7 and TLR9 on these cells causes the rapid expression of this cytokine. 59,72 Interestingly, pDCs may contribute to a positive feedback loop in SLE patients (Fig. 2). Specifically, pDCs do not have the capacity to bind DNA and RNA autoantigen directly but instead require incorporation of these autoantigens into an immune complex. Upon immune complex binding to surface FcγRII on the pDC, the nucleic acid component of the complex stimulates TLR7 or TLR9, resulting in IFNα expression. 10,73 IFNα subsequently stimulates autoreactive B cells to proliferate, make more autoantibody, and isotype class switch (Fig. 1), thus contributing to the formation of additional immune complexes. 55,70 In addition, pDCs process and present autoantigen to autoreactive T cells, further promoting their activation and proliferation (Fig. 1).
Myeloid Dendritic Cells Contribute to the Pathogenesis of SLE
Like pDCs, myeloid dendritic cells (mDCs) appear to participate in the pathogenesis of SLE. In mice, mDCs take up DNA and RNA immune complexes in an Fc-dependent manner and the nucleic acid component signals through TLR7 and TLR9, 32 respectively, thus activating the cell (Fig. 1). The mDCs subsequently process and present autoantigen to autoreactive T cells, further amplifying the autoimmune T-cell response. 54 Propagating the autoimmune antibody response, mDCs simultaneously produce the B-cell activating factor BAFF, which promotes B-cell activation and survival and promotes additional autoantibody production. 8
Autoreactive B-cell Activation by BAFF and TLR Ligands May Substitute for T-cell Help During Disease Progression
Although initial autoantibody production appears to depend on the interaction of autoreactive B and T cells, there is now evidence that as SLE progresses, autoreactive B cells can produce class-switched antibodies in the absence of T-cell help. Rather, B cells can be directly activated by a combination of signals, including TLR 7/9 recognition of nuclear antigen–antibody complexes and BAFF binding to the BAFF receptor TACI (Fig. 2D). In support of this mechanism, BAFF-transgenic mice produce autoantibodies and develop nephritis and other SLE-like symptoms. 44,77 Interestingly, when these mice are crossed with a T cell–deficient strain, the BAFF-transgenic T cell–deficient progeny still develop disease and produce abundant autoantibodies of the IgM and IgG but not IgA isotypes, proving that autoantibody production can occur in the absence of T cells. 44 Production of autoantibodies in this model was shown to depend on signaling through TLR 7/9 because BAFF-transgenic mice that are reconstituted with MyD88-deficient B cells (which have defective TLR 7/9 signaling) do not develop disease.44 Further studies have demonstrated that signaling through autoreactive B cell TLR 7/9 upregulates the BAFF receptor TACI, thus rendering the cells responsive to BAFF. 44,87,118 Thus, as disease progresses, autoantibody production may occur independent of T-cell help via a loop involving TLRs, BAFF, and IFNα (Figs. 1, 2).
Autoantibody–Antigen Immune Complexes Contribute to Tissue Damage
The positive feedback loop serves to provide ever-increasing amounts and diversity of autoantibody and activated leukocytes that contribute to the clinical manifestation of disease. Nephritis is the most common lesion in SLE patients because autoantibody–antigen immune complexes in circulation are deposited in the walls of glomeruli during filtration. 129 Additionally, autoantibody may recognize autoantigen trapped in the glomerular basement membrane 84 or cross-react directly with basement membrane components. 129 The infiltration of Fc-bearing leukocytes is important for the development of nephritis because NZB/W mice deficient in Fc receptors are protected from developing renal disease despite the presence of complement. 11,24 The notion that complement is not essential for the development of lupus nephritis is supported by the observation that C3-deficient MRL/lpr mice still develop renal disease. 104 Regardless, the infiltrating leukocytes become activated, produce cytokines, and promote mesangial cell proliferation. In addition to being deposited in the glomerulus, autoantibody–immune complexes can be deposited in the blood vessels of other tissues and/or the epidermal basement membrane, 45 thus promoting leukocyte recruitment and activation, complement activation, and local inflammation.
SLE in Other Species
Although the discussion to this point has focused on SLE in mice and humans, spontaneous disease is recognized in other species, including rats, cats, dogs, rabbits, guinea pigs, pigs, hamsters, Aleutian minks, horses, and primates. 1,2,40,41,47,58,75,110,124 Of these species, the disease has been most carefully characterized in the dog. Below we describe the characteristics of SLE in several domestic species and discuss the practical limitations of using larger animal models in SLE research.
Like the disease in humans, SLE in the dog is a chronic illness characterized by periods of progression and remission. Clinical symptoms in the dog commonly include polyarthritis (91%), glomerulonephritis (65%), mucocutaneous lesions (60%), lymphadenopathy, and splenomegaly. 39 Skin lesions can be systemic or localized, typically to the nose (eg, discoid SLE). 43,46 Less common manifestations of SLE in the dog include hemolytic anemia, thrombocytopenia, thrombosis, 112 and complete heart block. 78 Antibodies against single-stranded DNA, double-stranded DNA (dsDNA), and histone proteins are present in 2%, 21%, and 71% of dogs, 82 and anti-soluble nuclear antigen, anti-Sm antigen, and anti-ribonucleoprotein antibodies are present in 74%, 24%, and 10% of dogs, 25 respectively.
Spontaneous SLE rarely occurs in nonhuman primates and is characterized by the presence of antinuclear antibodies, hemolytic anemia, membranoproliferative glomerulonephritis, and arthritis. 2 Also, an SLE-like disease can be induced in nonhuman primates by feeding the animals alfalfa seeds or L-canavanine sulfate, a constituent of alfalfa sprouts. 79
SLE has been recognized as a spontaneous disease in cats, characterized by glomerulonephritis (45%), neurologic symptoms (41%), arthritis (41%), anemia (36%), and dermatologic disease (32%). 47,75 Interestingly, treatment of cats with propothiouracil induces the production of anti-DNA and anti-myeloperoxidase antibodies, as well as hemolytic anemia. 6,123
Horses rarely develop SLE, and the disease is characterized by weight loss, polyarthritis, proteinuria, thrombocytopenia, bilateral symmetric alopecia, seborrhea, oral ulceration, and lymphadenopathy. 40,122 Abnormal laboratory findings include a Coombs-positive hemolytic anemia and positive antinuclear antibody test result. Affected skin is characterized by interface dermatitis with linear deposition of IgG at the basement membrane zones of the epidermis and hair follicles. 40
Although mouse models are typically used to study the pathogenesis of SLE for practical reasons, attempts have been made to establish large animal models of the disease. For example, immunization of dogs with heparin sulphate induces an SLE-like disease characterized by the development of antinuclear antibodies, proteinuria, and skin lesions, including deposition of IgM and C3 at the dermo-epidermal junction. 20 Likewise, rabbits immunized with purified Sm and small nuclear ribonucleoproteins produced precipitating and hemagglutinating antibodies against the antigens. 52 Also, female baboons immunized with a peptide from the Sm B/B’ spliceosomal protein developed antibodies to multiple regions of the Sm B/B’ protein, as well as reactivity against other spliceosomal proteins. Additionally, evidence of epitope spreading was observed in the baboons because, in addition to producing antibodies to the protein to which they were immunized, they produced antinuclear ribonucleoprotein and anti-dsDNA antibodies. 4 One major issue with studying SLE in large animals is that the populations are outbred and this variable genetic background often results in inconsistent manifestation of disease. Furthermore, the expense of caring for large animals and the relatively large quantity of drug required for efficacy studies (as compared to those for a mouse) limit the practical use of these models.
Toward Effective Therapy for SLE
As previously mentioned, genetic and functional abnormalities contributing to the pathogenesis of SLE are many and varied. As these various abnormalities culminate in a complex positive feedback loop, effective therapy may require that one or more steps of the loop be severed. A variety of approaches have been attempted over the years.
Generalized Immunosuppressants and Cytokine Blockade
The least specific approach to treat SLE is to induce a state of generalized immunosuppression. Drugs used in this approach include cyclophosphamide/fludarabine, methotrexate, tacrolimus, mycophenolate mofetil, azathioprine, and corticosteroids (reviewed in Kuiper-Geertsma and Derksen 66 ). Even the immunomodulatory drugs lenalidomide (Revlimid; Celgene Corporation, Summit, NJ) 108 and thalidomide 14 are being evaluated for the treatment of lupus. The major disadvantage of using generalized immunosuppressants is that the patient is more susceptible to bacterial or fungal infections. Because generalized immunosuppression is undesirable, attempts are underway to inhibit specific cytokines that may propagate the positive feedback loop. Clinical trials are in progress to evaluate blocking specific cytokines, including TNF-α, 5 IFNα, 127 BAFF (belimumab), and interleukin-6 (tocilizumab) (reviewed in Aran and Putterman 3 and Wentworth and Davies 126 ).
Innate Immune Inhibitors
Because the innate immune system is essential to the pathogenesis of SLE, inhibitors of innate responses may be effective at controlling the disease. Serendipitously, SLE patients have been treated with chloroquine for years, and the mechanism of action remained unknown until recently. Chloroquine prevents the acidification of endocytic vesicles, a step that is critical for signaling through endosomal TLR7 and TLR9 when they are engaged with DNA- and RNA-containing immune complexes, respectively. 115 Various TLR inhibitors are being evaluated in preclinical models and in the clinic. 115
Adaptive Immune Inhibitors
B cells are considered to be the main culprits in SLE by virtue of their production of autoantibody, as well as their direct activation of autoreactive T cells and sensitization of other APCs to autoantigen. Thus, one might predict that depletion of B cells is an effective treatment for human SLE patients. Indeed, depletion of CD20+ B cells with rituximab in SLE patients leads to clinical remission 107 and reduced autoreactive CD4+ T cells in circulation. An antibody designed to deplete CD22+ B cells is in clinical development. 126 Promising results are emerging with a monoclonal antibody against BAFF (anti-BLys; belimumab) in phase 3 clinical trials in seropositive patients with SLE (S. Navarra, unpublished data, American College of Rheumatology annual meeting, 2009). Treatment with belimumab reduces the number of circulating B cells and the titer of anti-dsDNA antibodies (D. J. Wallace, unpublished data, American College of Rheumatology annual meeting, 2006; European League Against Rheumatism annual congress, 2006).
Costimulation Inhibitors
As mentioned previously, autoreactive T and B cells stimulate one another to initiate and propagate the feedback loop in SLE. Accordingly, blockade of the various costimulatory pairs, including CD28/CD80, CD28/CD86, 37 and ICOS/B7RP-1, 53 as well as TNF superfamily members such as CD40/CD40L, 31 have profound benefits in mouse models of SLE. Similarly, clinical trials to evaluate the efficacy of CD28/CD80 and CD28/CD86 with CTLA4-Ig (abatacept) are currently underway (reviewed in Wentworth and Davies 126 ).
Conclusions
SLE is a complex disease involving dysfunction of the immune system at multiple levels. Years of painstaking research with mouse models and human patients have helped us understand the pathogenesis of this disease. A variety of genetic abnormalities and environmental factors contribute to the onset and progression of SLE, and a different combination is likely responsible in each patient. As our knowledge of this disease continues to increase, we may be able to identify those pathways and/or immune responses that are aberrantly functioning in each patient and so design a personalized course of treatment to maximize clinical efficacy and minimize the undesired side effects associated with nonspecific immune suppression.
Footnotes
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.