
Abstract
Age-related macular degeneration (AMD) is a degenerative condition that begins in Bruch’s membrane and progresses to involve the retinal pigment epithelium and ultimately the overlying photoreceptors. The only required etiologic factor is age, and AMD is regarded as the leading cause of blindness in individuals older than 65 years. AMD results from variable contributions of age, environment, and genetic predisposition. Many loci are linked to AMD; in the majority of cases, the disease is associated with polymorphisms within these genes, rather than mutations that ablate gene function. The etiologic complexity of AMD is reflected by the paucity of animal models that entirely replicate the human disease. This review compares the salient anatomy of the primate and rodent retina, particularly in the light of AMD pathology. It next discusses prevailing hypotheses explaining how AMD may develop. These include the role of complement activation and macrophage chemotaxis in AMD, molecular mechanisms of choroidal neovascularization, and the roles of oxidative damage and lipid metabolism. Finally, the article gives an overview of spontaneous and induced nonhuman primate models and describes relevant mouse models in the context of each pathogenetic mechanism.
Age-related macular degeneration (AMD) is a progressive condition of the retinal pigment epithelium (RPE), its supporting basement membrane, and the overlying photoreceptor layer. It affects the macula, the central area of the retina responsible for high-acuity daylight vision. Among individuals older than 65 years, AMD is regarded as the leading cause of blindness in the industrialized world. 39
AMD has no single cause and results from variable contributions of age, genetic predisposition, and environment. Established epidemiologic risk factors include cigarette smoking, diet, female sex, Caucasian race, and a family history of AMD. 52,60,61 Because AMD is rare in individuals younger than 55 years, the only required risk factor is age, which implicates the multitude of cellular changes that accompany normal aging in the pathogenesis of AMD. The challenge is to identify how these are altered to tip the process toward overt disease.
With age, most people accumulate small, well-demarcated deposits of extracellular material beneath the RPE. 5,45 These are known as small, hard drusen and are not pathogenic. However, if these deposits enlarge, they become associated with regional RPE loss and eventual degeneration of overlying photoreceptors. These changes are funduscopically visible, and their progression is used to classify lesions along the continuum of early to late dry AMD. 18 The pathogenetic mechanisms of this process center on inflammation, oxidative damage, and RPE senescence 45 and are predominantly modeled in the mouse.
In the minority of cases, late AMD is accompanied by growth of blood vessels from the choroid through Bruch’s membrane toward the retina. 36 These vessels may bleed, resulting in acute loss of central vision. Fortunately, this wet form of AMD is the one for which significant palliative treatments have been developed. Rodents and nonhuman primates are the primary models in which choroidal neovascularization is induced and antiangiogenic therapies are tested.
The etiologic complexity of AMD is reflected by the relative paucity of effective therapies, preventive strategies, and good animal models in which to study it. This is in part due to anatomical differences in animal and primate retinas, as well as the protracted time needed for the disease to develop. The following review describes the most commonly used animal models of AMD. It begins with a comparative overview of the salient anatomy across species, as well as how AMD affects each anatomical compartment. Next, prevailing hypotheses explaining how AMD may develop are discussed, and relevant animal models are described in the context of these pathogenetic mechanisms.
Comparative Retinal Anatomy and the Pathology of AMD
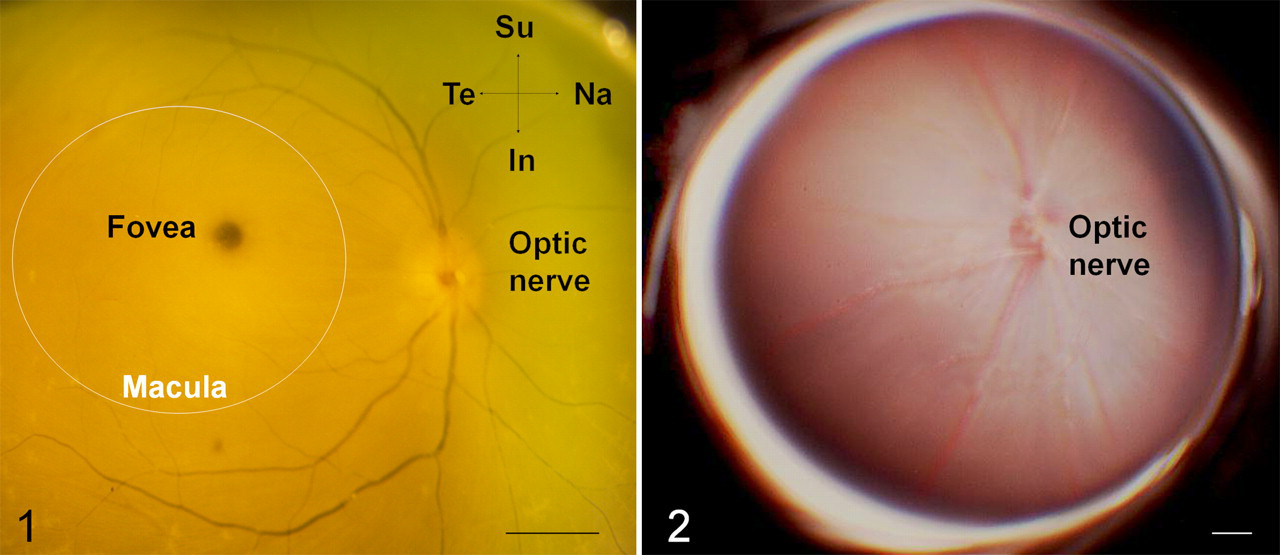
AMD affects the macula, a cone-dense region located just temporal to the optic disc (Fig. 1 ). The macula mediates high-acuity central vision and is present in primates, as well as some birds and reptiles. 87 At its center is the fovea, a cone-dominated avascular region of maximal visual acuity. The mouse lacks a macula (Fig. 2 ) and has a much lower proportion of cones in its retina. Nevertheless, the mouse is the most commonly utilized animal model of AMD. This is the case for 2 reasons. First, the histologic anatomy of the primary site of AMD (RPE, Bruch’s membrane, and choriocapillary layer) is well preserved across species. Second, the mouse is genetically manipulable and is thus the most practical model in which to test the numerous genetic alterations associated with AMD.
The Retinal Pigment Epithelium
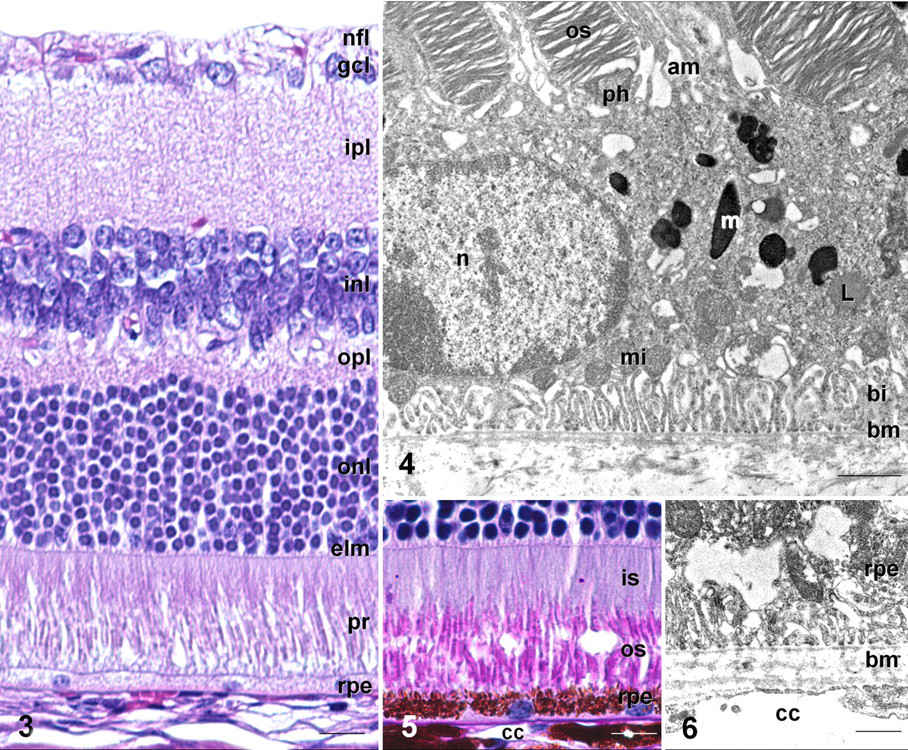
The RPE is derived from neuroectodermal cells of the optic primordium 104 and so constitutes the outermost layer of the retina (Figs. 3–7). It performs a number of critical functions that, if disrupted, result in the death of photoreceptors. In the majority of chordates, the RPE is a monolayer of cuboidal cells with distinct apical and basal polarity (Fig. 4). The apical aspect of RPE cells is intimately associated with photoreceptor outer segments (Figs. 3–5). Photoreceptor outer segments are continually renewed from their base, and a major function of RPE cells is to remove oxidized outer-segment disc material. 104
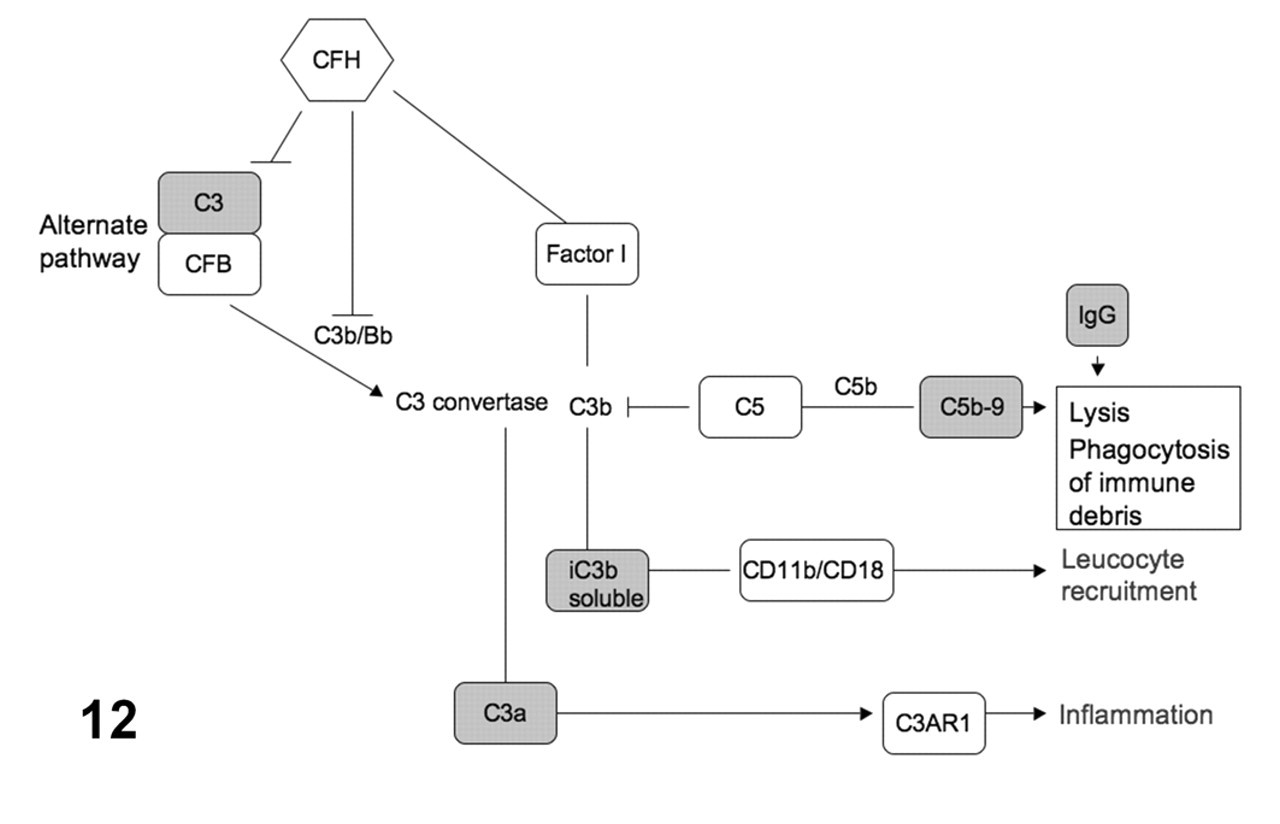
Complement pathway genes associated with age-related macular degeneration. Genes harboring polymorphisms associated with altered age-related macular degeneration risk are in large bold font. The classical, lectin, and alternative pathways converge on C3 through formation of protease complexes known as C3-convertases. These are able to cleave C3, generating C3b. C3b deposition promotes phagocytosis of structures upon which it is deposited; it can also cleave C5 and initiate the assembly of the membrane attack complex (C5b-9) that leads to complement-mediated lysis. The classical and lectin pathways converge through formation of C3 convertase (C4b2a). The alternate pathway begins with generation of C3b, either via the classical pathway or through exposure to antigenically foreign surfaces. Complement factor B (CFB) associates with C3b and generates C3 convertase (C3bBb) as well as C5 convertase through addition of another C3b molecule. Complement factor I regulates complement activation by cleaving C4b or C3b. Complement factor H (CFH) further inhibits complement activation by inhibiting association of C3b and CFB and enhancing CFI activity.
Microvilli that fringe the apical surface of RPE cells enclose and phagocytose the distal tip of photoreceptor outer segments (Fig. 4). These internalized discs are degraded and recyclable molecules (eg, retinal or docosahexaenoic acid) redelivered to photoreceptors. 7 Undegraded material accumulates in the lysosomal system as lipofuscin granules (Fig. 4). The principal component of lipofuscin is A2E (N-retinyledin-N-retinylethanolamin), a toxic by-product of the retinoid cycle. 101,116 Lipofuscin granules accumulate with age, and their contents, particularly A2E, are thought to contribute to RPE damage in AMD 116 (see next section).
In pigmented mouse strains, numerous ovoid melanosomes are present (Figs. 4, 5). With age, lipofuscin granules contain small melanin deposits, implicating some degree of autophagy. 114 Undigested intracellular material in aging RPE cells is removed via exocytotic activity. Partially digested membrane-bound proteins are disgorged by RPE cells and accumulate at the Bruch’s membrane, thus suggesting one source of drusen. 114
The basal cell membrane is characterized by distinct infoldings and abundant mitochondria (Fig. 4). The RPE delivers essential nutrients from the blood to the photoreceptors. 104 These include glucose, vitamin A (for regeneration of rhodopsin), and docosahexaenoic acid (for synthesis of photoreceptor outer-segment membranes). Deposition of drusen within Bruch’s membrane and between basal infoldings results in impaired chorio-RPE transport and contributes to degeneration of RPE and photoreceptors. 121
RPE damage is an essential feature of AMD. 45,121 Drusen deposition is accompanied by accumulation of lipofuscin within RPE cells, as well as their progressive atrophy and degeneration. The majority of murine models of AMD exhibit lipofuscin accumulation within RPE cells, or RPE pathology characterized by simultaneous loss and hypertrophy or hyperplasia of RPE cells.
A2E and Macular Predisposition
Phototransduction is initiated by light-induced isomerization of the 11-cis configuration of retinal (the visual pigment in opsin) to the light insensitive all-trans form. The RPE is required for cycling and reisomerization of retinal from the all-trans to the 11-cis form to regenerate visual pigments (the retinoid cycle). 104 One consequence of this process is the formation of A2E. A2E consists of 2 molecules of retinaldehyde (A) and 1 molecule of ethanolamine (E) and so forms in photoreceptor outer segments and in the RPE as a by-product of the retinoid cycle. 101 In the presence of light and oxygen, A2E generates free oxygen radicals and kills the cells in which it resides. A2E accumulates in lipofuscin granules over time, particularly in the macular region. 116 This may explain the predisposition of AMD to selectively affect this portion of the retina. Accumulation of A2E is a feature of AMD 116 as well as early-onset hereditary maculopathies such as the Stargardt macular dystrophies. It has been demonstrated in some mouse models of AMD, especially those that overlap with Stargardt macular dystrophy (see Mouse Models of Altered RPE–Photoreceptor Physiology section).
Bruch’s Membrane
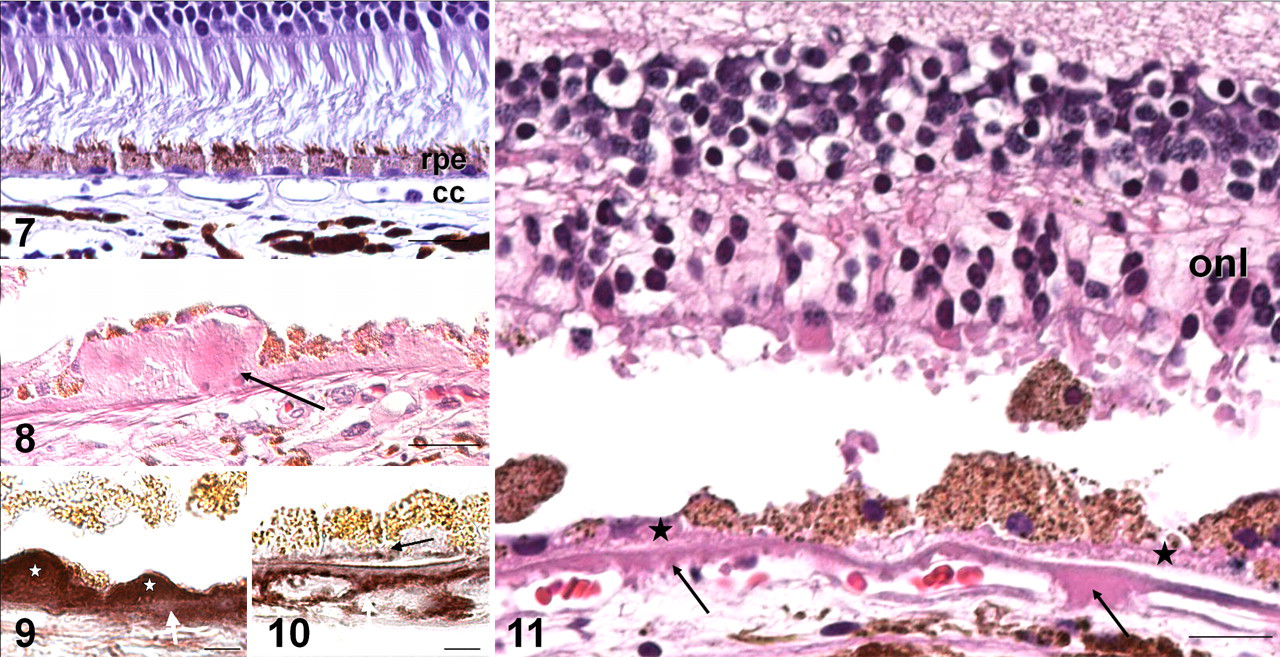
RPE cells rest upon the Bruch’s membrane, a trilaminar structure consisting of the basement membrane of RPE cells, an intervening elastocollagen layer, and an outer layer derived from the basal layer of the choriocapillaris (Fig. 6). This layering is more evident in primates than in mice. Normal aging is accompanied by thickening of the Bruch’s membrane and minor accumulation of small discrete sub-RPE deposits known as hard drusen within the macula. AMD is characterized by fragmentation and calcification of the Bruch’s membrane, as well as accumulation of soft drusen 42,103 (Figs. 8–11). Funduscopically, these are paler, larger, and poorly defined. Histologically, they exceed 63 µm in diameter and occur in 2 locations. Basal laminar deposits accumulate between the RPE basal plasma membrane and its basal lamina and contain long-spacing collagen. Basal linear deposits accumulate within the Bruch’s membrane and are principally composed of membranous debris. 42,103 By light microscopy, both types appear homogeneous and eosinophilic (Figs. 8, 11). As AMD advances, RPE cells degenerate to form confluent regions of hypopigmentation (geographic atrophy). Overlying photoreceptors also degenerate (Fig. 11), resulting in significant loss of central vision. This represents the most advanced stage of the so-called dry form of AMD.
The biochemical composition of drusen is complex 3,45,54 and includes complement components such as complement C3 and C5b-9 terminal complexes (Figs. 9, 10, 12), immunoglobulins, apolipoproteins B and E, fibrinogen, vitronectin, and amyloid β. Typical drusen are difficult to replicate in mice, although a variety of sub-RPE deposits are demonstrable in most models. In addition, many of the biochemical components of drusen can be demonstrated immunohistochemically.
The Choriocapillaris
The mammalian retina has 2 distinct circulatory systems. The inner retina is supplied by the retinal artery, which enters the eye via the optic nerve. The artery branches in the nerve fiber layer and ramifies to terminate in the inner plexiform layer. This vascular system is easily visualized on funduscopy (Figs. 1, 2, 13, 14), in contrast to the choriocapillary system, which is too fine to be normally visible.
The outer retina (photoreceptor and retinal pigment epithelial layers) is nourished by the choroidal circulation. This consists of the anterior and posterior ciliary arteries, derived from the ophthalmic artery. These vessels elaborate arterioles in the choroid and capillaries directly beneath the Bruch’s membrane (the choriocapillaris; Fig. 7). Accumulation of drusen within the Bruch’s membrane during AMD degrades this physical barrier and promotes its invasion by new vessels. This is termed neovascularization and is a complication in approximately 10% of advanced AMD patients. Because choroidal neovascularization results in serous detachment of the RPE or hemorrhage into the subretinal space, it is also known as wet AMD. Neovascularization occurs mostly commonly from the choroidal side, although in some cases, new vessels may extend from the inner retinal circulation and anastomose with choroidal vessels. This is termed retinal angiomatous proliferation, or chorioretinal anastomoses. 65 In cases of choroidal neovascularization, leakage from newly formed choriocapillary vessels can be visualized using fluorescein angiography (Fig. 14 ).
The RPE secretes a proangiogenic factor, vascular endothelium growth factor A (VEGF). 6 The role of an intact Bruch’s membrane is underscored by the inability of VEGF-overexpressing mice to develop choroidal neovascularization unless the Bruch’s membrane is breached. 80,95 The RPE also secretes the antiangiogenic pigment epithelium-derived growth factor (SERPINF1 or PEDF). PEDF expression is reduced in AMD eyes, 5 suggesting that the homeostatic balance between these 2 proangiogenic and antiangiogenic proteins is disrupted in AMD.
Mouse Models of Altered RPE–Photoreceptor Physiology
Three models are instructive in explaining how alterations of photoreceptor–RPE function may predispose to the pathology of AMD. The first two result from defects in photoreceptor proteins, whereas the last (based on the cathepsin D transgenic mouse) has its genetic alteration limited to the RPE. The models demonstrate that a variety of mechanisms that impede normal RPE-mediated degradation of photoreceptor outer-segment material ultimately result in pathology of the RPE, its basement membrane, and photoreceptors.
Abcr–/– mouse
ABCR (ATP-binding cassette transporter, retina specific)—otherwise known as ABCA4 (ATP-binding cassette, subfamily A, member 4)—is a transmembrane protein located on photoreceptor outer-segment discs. Its role is to facilitate removal of the all-trans form of retinal following photobleaching so that the RPE can regenerate the 11-cis form. 115 Mutations in this gene result in an array of retinal-degenerative disorders, including AMD, and an early-onset form of macular disease known as Stargardt macular dystrophy. 1 Both conditions are characterized by excessive lipofuscin accumulation in the RPE. Abcr–/– mice fail to regenerate light-sensitive 11-cis retinal and consequently display prolonged dark adaptation. In addition, they accumulate A2E 101,115 in rods and in the RPE, as reflected by markedly increased RPE lipofuscin in Abcr–/– mice and eventual degeneration of RPE and overlying photoreceptors.
Formation of A2E is completely inhibited when Abcr–/– mice are raised in darkness. These data suggest that strong light exposure may accelerate AMD and that patients may slow progression by limiting exposure to light. 71 Maiti et al demonstrated an alternative approach to limiting A2E accumulation in RPE cells. 67 RPE65 is an RPE-expressed protein that is required for regeneration of 11-cis retinal. Treatment of the Abcr–/– mouse with RPE65 antagonists prevents the formation of A2E. 67 These proof-of-principle studies are encouraging but not yet clinically applicable. A normal retinoid cycle is required for rod function, and loss of RPE65 is associated with its own blinding condition: Leber congenital amaurosis type 2. 70
ELOVL4 transgenic mouse
ELOVL4 transgenic mice 58 express a mutant form of human ELOVL4 (elongation of very-long-chain fatty acids-like 4), another protein associated with Stargardt macular dystrophy. ELOVL4 is located in rods and cones and is thought to promote synthesis of long-chain fatty acids in photoreceptor outer-segment discs. ELOVL4 transgenic mice develop increased RPE lipofuscin and excessive A2E, presumably through accumulation of indigestible disc membranes containing abnormally synthesized very-long-chain fatty acids.
Cathepsin D transgenic mouse
Rakoczy et al 85 generated transgenic mice (Ctsdmcd/mcd ) expressing an enzymatically inactive form of cathepsin D in RPE cells, which results in impaired processing of phagocytosed photoreceptor outer segments and accumulation of ROS material within RPE. By 11 months of age, increased lipofuscin is present in RPE cells, as accompanied by RPE cell loss, hyperplasia, and degeneration of overlying photoreceptors. Of particular interest was the deposition of amorphous material beneath RPE cells and their basal lamina. This model strongly suggests that sub-RPE deposits can result not only in the absence of choriocapillary pathology but from impaired degradative capacity of RPE cells with subsequent extrusion of undigested material across the basal laminae of RPE cells. This model supports the recent suggestion by Wang et al 114 that aging RPE cells disgorge partially digested membrane-bound proteins into the sub-RPE space.
The Genetics of AMD
Modeling AMD in mice is complicated by several factors. Multiple genes are associated with AMD risk in human families (Table 1 ). In the majority of cases, risk is associated with polymorphisms within these genes, rather than mutations that ablate gene function. Replicating these polymorphisms at the genetic level in mice requires transgenic expression of the human variant with ablation of the native mouse gene. Furthermore, AMD risk is influenced by the combinatorial effects of polymorphisms in several genes and is further altered by epidemiologic factors such as smoking. Duplicating these human susceptibility haplotypes in mice is a challenging task. In fact, many existing mouse models of AMD are the result of gene defects not associated with AMD in humans (Table 1). However, such models reveal aspects of RPE biology that have furthered understanding of AMD pathogenesis in humans. The following sections focus on 2 predominant areas of AMD genetics: first, CFH (complement component factor H) and the complement pathway; second, HTRA1/ARMS2. Table 1 provides a list of current known AMD loci, as well as an extensive list of mouse models, organized according to the pathophysiologic principles that constitute the major sections of this article.
Human ARMD Disease Loci and Animal Models a
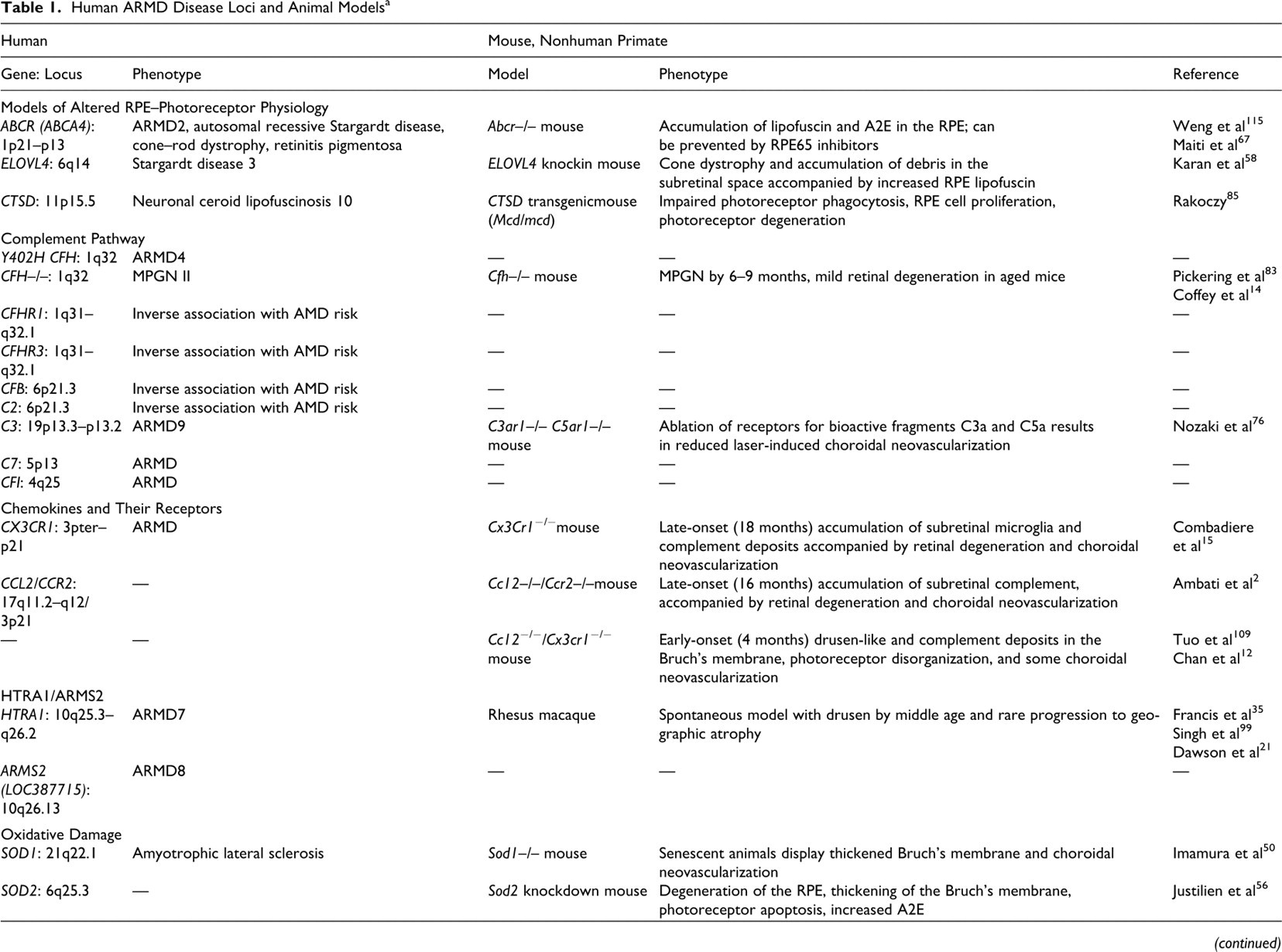
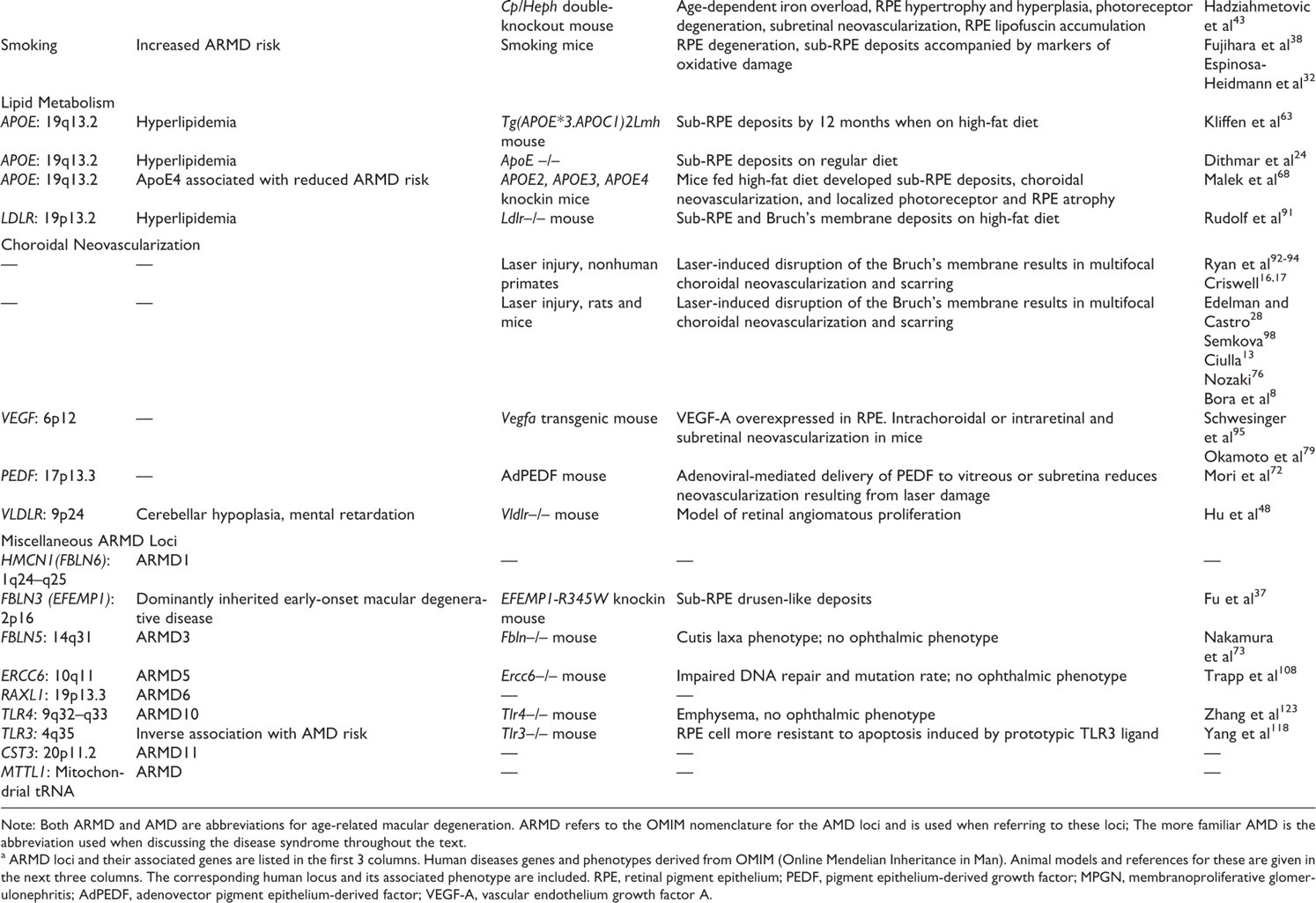
Note: Both ARMD and AMD are abbreviations for age-related macular degeneration. ARMD refers to the OMIM nomenclature for the AMD loci and is used when referring to these loci; The more familiar AMD is the abbreviation used when discussing the disease syndrome throughout the text.
a ARMD loci and their associated genes are listed in the first 3 columns. Human diseases genes and phenotypes derived from OMIM (Online Mendelian Inheritance in Man). Animal models and references for these are given in the next three columns. The corresponding human locus and its associated phenotype are included. RPE, retinal pigment epithelium; PEDF, pigment epithelium-derived growth factor; MPGN, membranoproliferative glomerulonephritis; AdPEDF, adenovector pigment epithelium-derived factor; VEGF-A, vascular endothelium growth factor A.
Inflammation in AMD
Inflammation has been proposed as a central mechanism in the pathogenesis of AMD. 45 Support for this hypothesis comes primarily from the many inflammatory constituents of drusen (including alternative pathway components). 54 In 2005, this hypothesis received support from genetic studies in AMD populations when a major risk allele for AMD, the Y402H polymorphism in complement factor H, was discovered. 28,47,61 This polymorphism accounts for approximately 50% attributable risk, and it focused attention on dysregulation of the complement pathway as a major pathogenetic mechanism in AMD.
CFH and Other Complement Pathway Genes
Evidence from human and animal studies suggests a model of AMD pathogenesis in which the disease can arise from either increased complement deposition in the RPE and the Bruch’s membrane 45 or failure to remove activated complement components from these sites. 2
The complement system is a crucial component of innate immunity against microbial infection. 89 Its components are organized into 3 activation pathways: the classical, lectin, and alternative pathways. Upon activation, these pathways result in the formation of unstable protease complexes, named C3-convertases. These are able to cleave C3, generating C3b. C3b deposition promotes phagocytosis of structures upon which it is deposited; it can also cleave C5 and initiate the assembly of the membrane attack complex (C5b-9) that leads to complement-mediated lysis (Fig. 12 ).
The first complement associated gene polymorphism to be identified in AMD populations was the Y402H (tyrosine to histidine substitution at amino acid 402) variant of complement factor H (CFH). 28,44,47,62 Individuals homozygous for the risk-conferring 402H CFH variant have a five- to sevenfold greater risk of developing AMD. CFH is a negative regulator of the complement system, and it inhibits the alternative pathway by promoting factor I–mediated inactivation of C3b or by displacing factor Bb from the C3bBb complex. 124 Impaired CFH activity could conceivably result in overactivity of the alternate complement pathway, excessive complement deposition, and tissue injury in AMD. 44
This hypothesis has been difficult to prove. Complement components (Figs. 9, 10), CFH, and other inflammatory mediators are present in AMD lesions, 55,88 but precisely how the Y402H CFH polymorphism predisposes to AMD is unclear. Complete lack of CFH function does induce systemic activation of the complement system but results in glomerulonephritis, not AMD. 83 Individuals carrying the 402H risk variant of factor H do not invariably develop AMD, and those with the protective Y402 variant of CFH can still develop the disease, which implies that additional factors are required.
The Y402H polymorphism occurs in a region of CFH that binds to C-reactive protein (CRP), an acute-phase protein promoting complement activation. 100 Elevated CRP is a known cardiovascular risk factor, and it is associated with AMD. 96 Failure of CFH to adequately control CRP-dependent complement activation is a proposed mechanism by which the 402H variant of CFH promotes AMD. 113 A more likely mechanism is that AMD results from a risk haplotype (ie, inheritance of several AMD risk–associated polymorphisms in genes that control age-related inflammatory processes). This hypothesis is supported by identification of polymorphisms in genes encoding other complement pathway proteins. 23,82 For example, increased AMD risk is associated with polymorphisms in complement C3, 69,119 complement 7, 23 and complement factor I. 34 Polymorphisms in other genes are reported to reduce AMD risk; these include complement component B and complement component 2, 102 as well as the adjacent CFH-like genes CFHR1 and CFHR3. 49
The Cfh–/– mouse
Like humans lacking CFH function, the Cfh–/– mouse experiences systemic consumption of C3, deposition of C3a in the glomerular basement membrane, and glomerulonephritis, resulting in death by 12 to 15 months. 84 Animals that survive to 2 years develop mild loss of photoreceptors, reduced rod function, and increased deposition of complement in the photoreceptor layer. 14
Mice deficient in C3ar1–/– and C5ar1–/–
C3a and C5a are the bioactive fragments of C3 and C5 and are present in human drusen deposits in AMD. 3 Both can induce VEGF expression in vitro and in vivo, thus potentially promoting choroidal neovascularization. They affect their biological responses through their receptors, C3aR and C5aR. Nozaki et al 76 showed that genetic ablation of receptors for C3a or C5a reduces VEGF expression, leukocyte recruitment, and choroidal neovascularization formation after laser injury, implicating local complement deposition as a risk factor for progression of AMD.
Chemokines and Chemokine Receptors
AMD is characterized by not only complement activation but also accumulation of macrophages in the vicinity of drusen, as well as retinal microglial activation and accumulation. 54,59,81 AMD risk haplotypes have been identified in other another gene family associated with immune regulation—the chemokines and their receptors. 82 Chemokines constitute a family chemotactic cytokines that direct the migration of leukocytes during inflammation. They are divided into 2 major subfamilies, CXC and CC, based on the arrangement of the first 2 of the 4 conserved cysteine residues, and they function through their binding to a subset of 7-transmembrane, G protein–coupled receptors. 41 Attention has fallen on both chemokine subfamilies to explain how complement activation, macrophage kinetics, and AMD may be related.
Cc12–/– and Ccr2–/– mice
A mouse model provided one of the first clues that AMD pathology may result from impaired clearance of complement deposits, although the model’s gene defect has not yet been linked to human AMD. CCL2 (chemokine, CC motif, ligand 2; alternatively known as monocyte chemotactic factor 1) and its receptor CCR2 mediate macrophage recruitment from the circulation to tissues at sites of complement deposition. Here, macrophages phagocytosed and remove activated complement components. 66 Older mice (> 16 months) lacking either of the genes experience a relatively broad spectrum of AMD-like pathology, including drusen-like Bruch’s membrane deposits, RPE loss, choroidal neovascularization, and photoreceptor atrophy. 2 This is accompanied by increased RPE deposition of C3 and C5, and in these mice, pathology presumably results from defective complement clearance resulting from their macrophage chemotactic defect. Biochemical analysis revealed that A2E levels are similarly increased in these mice.
Cx3cr1−/− mice
CX3CR1 (chemokine, CX3C motif, receptor 1) is a G protein–coupled receptor found microglia, macrophages, T-cells, and astrocytes. When bound by its ligand, fractalkine (or CX3CL1), CX3CR1 mediates adhesion and migration of leukocytes to inflamed tissues. Single-nucleotide polymorphisms (SNPs) in the CX3CR1 gene confer increased risk of AMD. 12,15,109 CX3CR1-positive cells in drusen have been identified as microglial cells, thus implicating this cell type in the pathogenesis of AMD in human eyes. 11
In 18-month-old Cx3cr1-deficient mice, 15 photoreceptor degeneration and choroidal neovascularization are accompanied by subretinal accumulation of microglia. On funduscopy, animals exhibit drusen-like punctuate lesions that correspond to accumulation of lipid-bloated subretinal microglia. Microglia are bone marrow–derived mononuclear cells that gain entry to the neuroretina through the inner retinal vasculature. Retinal damage of any type induces microglial migration into the subretinal space, from where they enter the circulation. 122 Combadière et al 15 hypothesized that CX3CRr1 deficiency led to impaired microglial egress from the retina, facilitating their accumulation in the subretinal space, where they generate drusen-like deposits
Cc12−/−/Cx3cr1−/− murine model
A disadvantage of the Cc12/Ccr2 and Cx3cr1−/− models is that they take a long time (16 to 18 months) to express the AMD-like phenotype. Compound Cc12 −/− /Cx3cr1 −/− mice exhibit, at an early age of 6 weeks, funduscopically visible drusen-like lesions, as well as histologically evident thickening of the Bruch’s membrane, drusen, local RPE hypopigmentation and degeneration, photoreceptor disorganization and atrophy, and choroidal neovascularization in some mice. 11,109 Increased complement deposition has been demonstrated in these areas. 90 These mice also exhibit accumulation of lipofuscin granules, as well as increased A2E accumulation, thus replicating these findings in human AMD (see earlier section, A2E and Macular Predisposition).
HTRA 1 and LOC387715/ARMS2 in AMD
A second major AMD-susceptibility locus is present on 10q26, where SNPs that span a 200-kb region show strong association to AMD. Two SNPs are most strongly linked to AMD. The first, rs11200638, identifies a promoter variant in the HTRA1 gene as a major risk factor for developing wet AMD. 22,117 The HTRA1 gene encodes a member of a family of widely expressed serine proteases. 78 Its expression in human fibroblasts increases with advancing age. 54 HTRA1 appears to regulate the degradation of extracellular matrix proteoglycans. This activity is thought to facilitate access of other degradative matrix enzymes (eg, collagenases and matrix metalloproteinases) to their substrates. 40 Conceivably, overexpression of HTRA1 may alter the integrity of the Bruch’s membrane, favoring the invasion of choroidal vessels across the extracellular matrix. HTRA1 also binds and inhibits transforming growth factor-β, an important regulator of extracellular matrix deposition and angiogenesis. 78
Transfection of RPE cells with plasmids bearing the risk-promoting HTRA1 sequence resulted in increased HTRA1 secretion. 22 This finding, with immunohistochemical evidence of HTRA1 in drusen of AMD patients, 117 suggests that the sequence change in the HTRA1 promoter enhances the transcription of HTRA1 in individuals homozygous for the risk allele. Additional studies 9,64,120 have also found the strongest association between AMD and the HTRA1-associated SNP.
These data contrast with other reports in which a closely located SNP (rs 10490924) in the coding region of a simian-specific mitochondrial protein, LOC387715/ARMS2, is most strongly associated with AMD. 53,57,86 Currently, it is unclear whether HTRA1 and LOC387715 are causative in AMD. Their identifying SNPs are so close that their differential association with AMD depends on the rare recombinant individual within the intervening 6-kb region that separates them. This dilemma may be solved using nonhuman primate linkage studies. Recent preferential association of HTRA1 in rhesus macaque colonies supports the involvement of this gene in AMD pathogenesis (see Spontaneously Occurring Primate Models of AMD section).
Oxidative Damage and AMD
Although inflammation has received much attention, the long-standing theory that oxidative damage plays an important role in AMD continues to receive support. Apart from age, smoking is the most strongly associated risk factor for AMD 62 and may damage the retina by inhibiting generation of antioxidants (eg, plasma vitamin C, carotenoids), generating reactive oxygen species, and reducing choroidal blood flow. Furthermore, the only known intervention that can reduce AMD progression is consumption of antioxidant vitamins, lutein (a protective pigment within the retina), and zinc. 33 Several mouse models illustrate the capacity of oxidative damage to induce AMD-like pathology, although none of the associated genes are linked to typical AMD.
Sod1–/– mice
Mice deficient in Cu,Zn-superoxide dismutase (Sod1) express markers of oxidative stress in the RPE. 50 This is accompanied by age-related (> 10 months) accumulation of sub-RPE deposits, thickened Bruch’s membrane, and choroidal neovascularization. These lesions are worsened by exposure to excess light.
Sod2 Knockdown Mouse
Adeno-associated virus–mediated ribozyme delivery targeting the Sod2 message results in progressive degeneration of the RPE, thickening of the Bruch’s membrane, and progressive degeneration of the photoreceptor outer nuclear layer. 56
Ceruloplasmin/Hephaestin Double Knockout Mice
Iron export from cells is facilitated by the multicopper ferroxidases ceruloplasmin and hephaestin. 46 Patients with aceruloplasminemia experience accumulation of iron in some tissues, including the retina, and develop macular degeneration in middle age, 25 suggesting that iron accumulation may play a role in this disease. Mice deficient in both ceruloplasmin and hephaestin 43 display age-dependent iron overload, RPE hypertrophy and hyperplasia, lipofuscin accumulation, subretinal neovascularization, and photoreceptor degeneration. These changes peak at 12 months and are accompanied by indicators of oxidative stress and complement activation.
Cigarette Smoke
Smoking is the most significant modifiable risk factor 52,60 for AMD. Mice exposed to cigarette smoke display markers of oxidative damage accompanied by RPE loss and accumulation of sub-RPE deposits. 32,38
Lipid Metabolism and AMD
Several studies have reported an association between AMD and cardiovascular risk factors such as atherosclerosis and increased body mass index 111,113 as well as high-fat diet. 97 Based on many of these results, association between apolipoprotein E alleles and AMD risk has revealed conflicting results. Most recently, a statistical review article of these results concluded that APOE2 alleles are associated with increased risk but that APOE4 alleles are associated with protection. 106 The majority of mouse models that explore the role of lipid metabolism develop relatively modest sub-RPE deposits and Bruch’s membrane thickening.
APOE 3–Leiden Transgenic Mice: Tg(APOE*3.APOC1)2Lmh
These mice carry a dysfunctional atherosclerosis-associated form of the human APOE3 allele. 63 When placed on a high-fat diet, these mice develop sub-RPE deposits by a year of age.
ApoE-Null Mice
These mice experience hypercholesterolemia and develop thickening of the Bruch’s membrane and accumulation of sub-RPE deposits. 24
Targeted APOE Replacement Mice
These mice were developed expressing human APOE2, APOE3, or APOE4 alleles. 68 When maintained on a high-fat diet, these mice show APOE isoform-dependent AMD-like pathology, such as diffuse sub-RPE deposits, thickened Bruch’s membrane, and atrophy of the RPE. However, in contrast to genetic data in human AMD studies, 106 the data in this model indicates that APOE4 mice are the most severely affected, thus preventing a clear mechanistic model for how APOE alleles promote AMD.
Ldl receptor−/− Mice: LDL Receptor
On a high-fat diet, these mice develop lipid accumulation within the Bruch’s membrane. 91
Spontaneously Occurring Primate Models of AMD
Macular degeneration occurs in rhesus and cynomolgus macaques. In both, retinal lesions are characterized by drusen accumulation in the central retina. However, onset of fundus changes, and inheritance patterns differ in these 2 primate species. Macular degeneration in the cynomolgus macaque appears to be a model for early-onset maculopathies rather than AMD. In contrast, adult-onset macular degeneration in rhesus macaques resembles human AMD. Genetic studies in rhesus macaques may resolve which of two closely linked adjacent genes on human 10q26 (HTRA1 and LOC387715/ARMS2) are responsible for the disease.
Early-Onset Macular Degeneration in Cynomolgus Monkeys (Macaca fascicularis)
Early-onset macular degeneration with autosomal-dominant inheritance occurs in a colony of cynomolgus monkeys at the Tsukuba Primate Center. 74,75,105,110 Symptoms appear at 2 years and progress slowly throughout life. Fine, yellowish white dots within the macula are evident by funduscopy. On histology, affected animals have increased lipofuscin accumulation within RPE. Various-sized complement (C5) immunoreactive drusen are present between the RPE and choriocapillaris in the macular region.
Young age of onset and inheritance pattern in this model are reminiscent of a number of early-onset maculopathies in humans, rather than AMD. One of the first genes discovered for AMD was ABCA4, 1 the causative gene for Stargardt macular degeneration 1. Heterozygous carriers for this gene defect are at risk for AMD, suggesting that early and late maculopathies may share common pathophysiologic elements. Umeda et al 110 assessed the contribution of early-onset human maculopathy susceptibility genes in this cynomolgus colony and found no association between the monkey phenotype and 13 distinct human loci.
Adult-Onset Macular Degeneration in Rhesus Monkeys (Macaca mulatta)
Cayo Santiago, a small island off the east coast of Puerto Rico, is home to a closed colony of rhesus macaques. Funduscopic examination of these animals revealed macular drusen in approximately half of animals older than 9 years and all animals older than 25 years. 20,21,29 –31 Histology and ultrastructural studies confirmed the presence of small and large drusen within the Bruch’s membrane and beneath the RPE. 19 A small breeding colony derived from these animals was established at the University of Florida in 1994 and so exhibits similar findings. 19 Despite the much higher prevalence of macular drusen in young adult to middle-age monkeys compared to that in humans, the disease appears to be less progressive, with the rare animal progressing to geographic atrophy or choroidal neovascularization. 20,21,31
In contrast to early-onset macular degeneration in cynomolgus macaques, the inheritance pattern in rhesus macaques appears to be complex. Francis et al 35 genotyped a population of rhesus macaques at the Oregon National Primate Research Center. In these animals, a susceptibility locus in the rhesus homologue of human 10q26 was associated with macular phenotype. This locus is an important one in humans, and it contains 2 adjacent genes, LOC387715/ARMS2 and HTRA1. Both are transcribed in rhesus and human retina and RPE, and both contain sequence variants significantly associated with affected status in humans and macaques. As expected, the LOC387715/ARMS2 gene is present in simians only, thus corresponding with development of the macula. Similar to that of humans, a promoter polymorphism in HTRA1 that results in overexpression of the protein was identified in this colony in functional in vitro studies. A subsequent study of rhesus colonies from the Caribbean Primate Research Centre and the German Primate Centre duplicated these findings. 99 In contrast to human studies, this study indicated that only the promoter polymorphism in HTRA1 appears to be significantly associated with the disease phenotype.
Modeling Choroidal Neovascularization in Advanced AMD
Agents that retard neovascular growth and reduce subretinal hemorrhage represent a major development in therapy for advanced AMD. In rodents and nonhuman primates, neovascularization of the choroid can be induced following laser-induced rupture of the Bruch’s membrane. These lesions can subsequently be targeted by therapeutic agents. More recently, genetically altered mice have been used to explore angiogenic homeostasis within the RPE–choroidal interface, particularly the interaction between proangiogenic VEGF and antiangiogenic PEDF. The interface of these 2 approaches has brought several antiangiogenic agents into clinical use. These currently focus on inhibiting VEGF, 112 although additional use of PEDF via gene therapy is likely in the future. 10
Laser Injury in Nonhuman Primates
Laser photocoagulation ruptures the Bruch’s membrane and evokes a fibrovascular proliferative response that originates in the choroid. This response is the basis for modeling choroidal neovascularization in late-stage AMD and was developed in rhesus and cynomolgus macaques by Stephen Ryan in the late 1970s. 92 –94 More recently, the technique has been modified for use in the squirrel monkey. 16
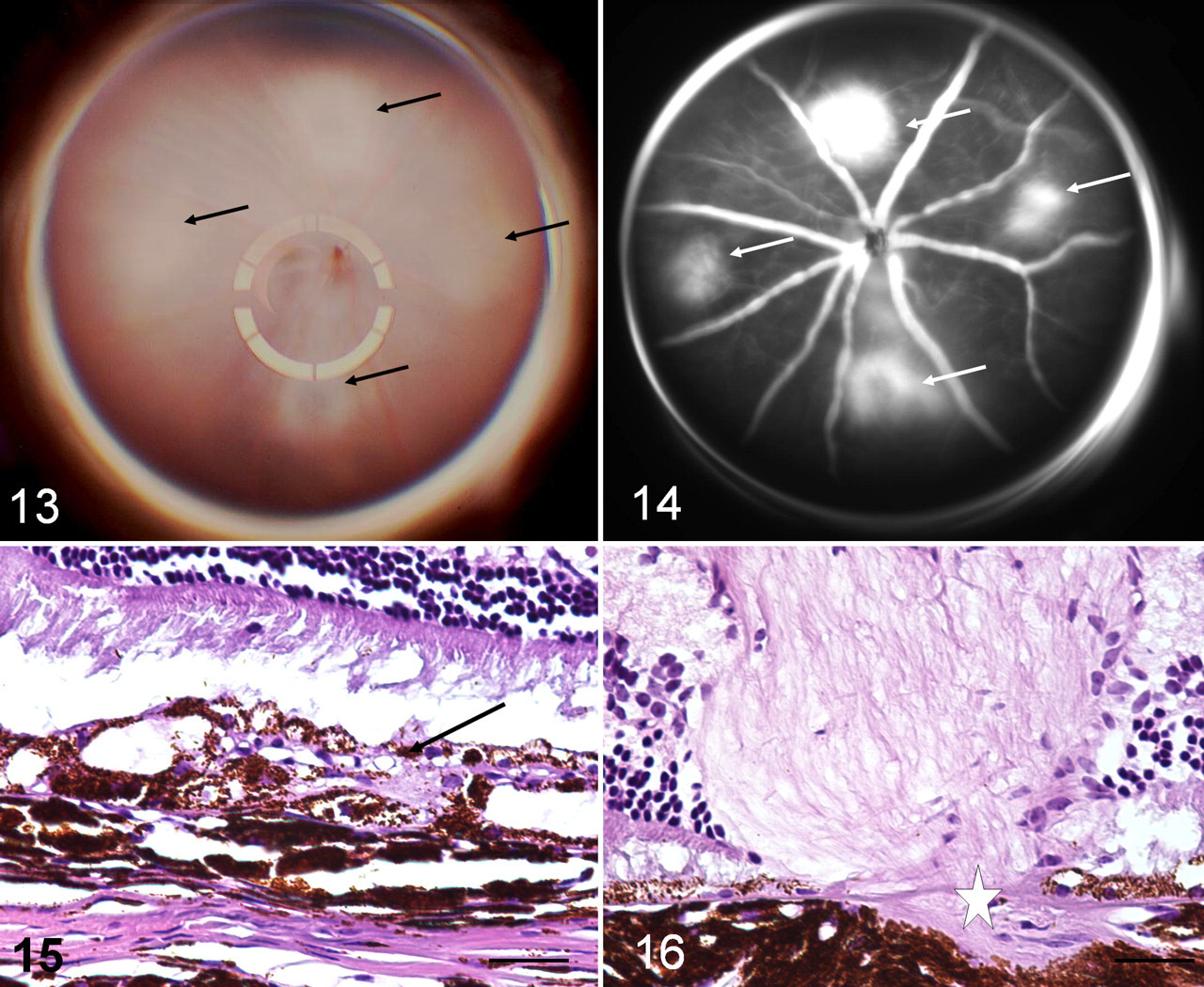
Using an argon laser, Ryan established the method currently in use today: to induce small spots of photocoagulation in a reproducible grid pattern in the temporal retina. The capillary-free zone of the fovea is avoided. 92 Spots are kept small and induced with sufficient power to rupture the Bruch’s membrane. This is funduscopically visible as a bubble at the time of photocoagulation. Photocoagulation induces thrombosis of choroidal vessels followed by re-endothelialization 48 hours later and growth of new vessels into the subretinal space by a week. 51 Because newly formed vessels are more permeable, neovascular development can be monitored with fluorescein angiography to assess vessel leakage. By this criteria, choroidal neovascularization peaks at approximately 2 to 4 weeks after photocoagulation. Experimental agents are typically given at approximately 1 month after injury, before vessels begin to resolve. 16,17 Spontaneous neovascular involution (indicated by decreased fluorescein leakage) commences at approximately 3 to 7 weeks and then gradually progresses (over a period of approximately 2 to 13 months) until leakage is no longer apparent at the site. 77,94 The extent of new vessel growth compared to poorly vascularized scarring can be variable in all models (Figs. 15, 16 ) and is influenced by species, location of injury in the retina, and intensity of the laser beam. 16,17 In some cases, laser injury can result in anastomotic vessels between choroidal and retinal circulations. 17 This lesion, known as retinal angiomatous proliferation, can be present as part of advanced AMD in humans, and it holds a poor prognosis.
Following sacrifice, tissues are usually examined with paraffin histology. Because these studies usually have a significant quantitative component, care must be taken during processing so that pathology of each laser site can be correlated with its in vivo angiographic progression.
Laser Injury in Rodents
Laser-induced neovascularization can be achieved in mice 8,26,76 and rats. 13,27,98 Compared to nonhuman primates, fewer laser sites (Fig. 13 ) are induced with lasers of shorter duration and lower power than that used in primates. Compared with that of primates, a larger proportion of lasered sites in rodents develop neovascularization. 27 Progression of new vessel growth is followed using fluorescein angiography (Fig. 14). Postmortem analysis is usually done with histology (Figs. 15, 16) but includes techniques such as RPE/choroid whole mounts. 27
Genetically Altered Mouse Models of Angiogenesis
Both VEGF (which is proangiogenic) and PEDF (SERPINF1; which is antiangiogenic) are expressed in the RPE–Bruch’s membrane–choriocapillaris complex. 4 In tissues from AMD patients, PEDF is significantly lower in RPE cells, the Bruch’s membrane, and the choroid than in control eyes. These data suggest that a decrease in PEDF may disrupt the balance of these 2 factors and create a proangiogenic environment that promotes choroidal neovascularization. 4,107
This hypothesis has received support from mouse studies that indicated that localized overexpression of VEGF results in concomitant angiogenesis and that PEDF overexpression protects against laser-induced choroidal neovascularization. Transgenic expression of VEGF in RPE cells (using the RPE65 promoter) results in choroidal vascular permeability, leukocyte adhesion, and intrachoroidal neovascularization. This, however, does not penetrate the Bruch’s membrane 95 or result in RPE pathology. If the VEGF transgene is expressed in the photoreceptor layer (using the rhodopsin promoter), new vessels originate from the inner retinal vasculature within the retina and extend into the subretinal space, where they form clusters of vessels surrounded by RPE cells. 79 Adeno-associated virus–mediated delivery of PEDF to the subretinal or vitreous space results in significant reduction of neovascularization resulting from laser injury in mice. 72
Last, retinal angiomatous proliferation in AMD patients is a relatively uncommon form of neovascularization in which new vessels originate from the inner retinal vasculature and extend through the subretinal space to communicate with the choroidal vessels. This phenotype is replicated in the Vldlr–/– mouse 48 and results in disruption of RPE cells and the Bruch’s membrane, retinal–choroidal anastomosis, and subsequent photoreceptor degeneration.
Conclusion
AMD is a complex disease that, so far, is incompletely modeled in animals. Evidence from human molecular genetic and animal studies support the notion that altered homeostasis of a multitude of mechanisms responsible for normal photoreceptor–RPE physiology can precipitate the disease. Fortunately, the molecular physiology of many of these can be explored in animal models and, in some cases, even in those whose gene defects are not the primary causes of AMD in humans. The rapid surge of information yielded by these approaches over the last 5 years offers the promise of understanding this once inexplicable disease.
Footnotes
Acknowledgement
I thank Dr Lu Fang for the fundus images of mouse retina.
The author declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
This work was supported by the National Institutes of Health (No. K01 RR16090-01A2) and the Claude D. Pepper Older Americans Independence Center at Yale University School of Medicine (No. P30-AG21342 [National Institutes of Health / National Institute on Aging; Dr Mary Tinetti, principal investigator]).