
Abstract
Objective
Reperfusion therapy is essential for preserving cardiac tissue in patients with acute myocardial infarction. However, ischemia–reperfusion exacerbates cardiac damage. Vascular endothelial cell–derived small extracellular vesicles play a pivotal role in ischemia–reperfusion injury; although the underlying mechanisms remain poorly understood. Therefore, this study aimed to elucidate the mechanisms of vascular endothelial cell–derived small extracellular vesicles and investigate their potential therapeutic roles.
Methods
Small extracellular vesicles derived from vascular endothelial cells were isolated using ultracentrifugation, and their characteristics were confirmed using transmission electron microscopy, nanoparticle tracking analysis, and western blotting. Their effects on myocardial injury was evaluated in vivo using a mouse myocardial ischemia–reperfusion model, and their role was further examined in vitro using a cellular hypoxia/reoxygenation model. The key micro ribonucleic acid in small extracellular vesicles was screened via high-throughput sequencing, and its regulatory effect on cardiomyocyte apoptosis was verified in vitro through intervention experiments. The key gene was predicted using miRanda and TargetScan, and the related interaction was verified by a dual-luciferase reporter assay and ribonucleic acid immunoprecipitation.
Results
Our in vivo study revealed that small extracellular vesicles significantly attenuated ischemia–reperfusion-induced cardiomyocyte apoptosis and improved cardiac function. We also analyzed micro ribonucleic acid expression in small extracellular vesicles and found that miR410-3p was highly expressed and associated with cellular apoptosis. In vitro experiments demonstrated that small extracellular vesicles increased miR410-3p expression in cardiomyocytes and that miR410-3p effectively inhibited cardiomyocyte apoptosis in a cellular ischemia–reperfusion model. Conversely, miR410-3p inhibition attenuated the protective effect of small extracellular vesicles. Moreover, we found that SMAD family member 7 is one of the target messenger ribonucleic acids of miR410-3p and affects cellular apoptosis by modulating nuclear factor kappa-light-chain-enhancer of activated B cells. Cellular experiments verified that the antiapoptotic effect of miR410-3p was neutralized by SMAD family member 7 overexpression.
Conclusions
These results revealed that vascular endothelial cell–derived small extracellular vesicles attenuate ischemia–reperfusion-induced cardiomyocyte apoptosis via the miR410-3p/SMAD family member 7 axis. This study provides valuable insights into the molecular mechanisms underlying ischemia–reperfusion-induced myocardial damage and highlights potential therapeutic opportunities for mitigating it.
Keywords
Introduction
Acute myocardial infarction is caused by the sudden occlusion of a coronary artery and is a major cause of mortality worldwide. 1 Reperfusion therapy has become the primary strategy for reducing infarction-associated morbidity and mortality.2–6 Nevertheless, the reperfusion process has a latent propensity to exacerbate myocardial injury, resulting in a 50% increase in infarct size, a phenomenon known as ischemia–reperfusion (I/R) injury, 1 which can eventually contribute to ischemic heart disease.
Various pathophysiological mechanisms, including calcium and proton overload, oxidative stress, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, activation of apoptotic pathways, and myocardial hibernation, mediate the cellular response to I/R.7,8 The pathophysiological processes underlying cardiac I/R injury are complex.9,10 Cardiomyocyte apoptosis occurs at the onset of acute ischemic injury;11,12 however, the exact mechanism underlying this process remains unclear.
Small extracellular vesicles (sEVs) are a subtype of extracellular vesicles, typically with a diameter of 30–200 nm and contain biomarkers such as tumor susceptibility gene 101 (TSG101), cluster of differentiation (CD) 63 (CD63), CD81, and CD9. They are secreted by most cell types and enter the interstitial space and circulation.13–15 sEVs mediate nuanced effects on recipient cells through their cargo, including lipids, proteins, deoxyribonucleic acid (DNA), messenger ribonucleic acid (RNA) (mRNAs), and microRNAs (miRNAs).16,17 sEVs are associated with various physiological processes (e.g. cell–cell communication and secretion of active molecules) and the pathogenesis of numerous diseases (e.g. cancer, inflammatory diseases, and cardiovascular diseases).18–22 Additionally, they exhibit potential clinical applications. 23 Vascular endothelial cells (VECs) are emerging as prolific and pivotal contributors of sEVs and are involved in paracrine signaling that intricately shapes the microenvironment in cardiac reperfusion tissues. Despite increasing evidence supporting the central role of sEVs in modulating cardiomyocyte apoptosis, hypertrophy, fibrosis, and angiogenesis,24–28 few studies have investigated the intricacies of VEC-derived sEVs in I/R-induced myocardial injury.
This study investigated the role of VEC-derived sEVs in myocardial I/R injury. In vivo experiments demonstrated that VEC-derived sEVs attenuated myocardial I/R injury by reducing cardiomyocyte apoptosis. Mechanistically, miR410-3p emerged as a key contributor of VEC-derived sEVs, as identified via high-throughput sequencing. Furthermore, in vitro studies established miR410-3p as a primary mediator of sEV actions that inhibit cardiomyocyte apoptosis by modulating SMAD family member 7 (SMAD7). Overall, this study elucidated the role and mechanisms of VEC-derived sEVs in myocardial I/R injury and suggested that miR410-3p represents a multifaceted therapeutic target for alleviating I/R-induced myocardial injury.
Methodology
Isolation and identification of sEVs
VECs were purchased from the National Collection of Authenticated Cell Cultures (Shanghai, China) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; HyClone; USA) supplemented with 10% fetal bovine serum (FBS; HyClone; USA), 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco; USA). After reaching full confluence, the cells were incubated in FBS-free DMEM (Yeason; China) for 72 h to isolate sEVs. The sEVs were collected using a standard differential centrifugation protocol. 29 The culture medium (200 mL) was collected and centrifuged at 300 ×g for 10 min to remove dead cells. The supernatant was then centrifuged at 2000 ×g for 10 min and 10,000 ×g for 30 min to remove cellular debris and large vesicles, respectively. The resulting supernatant was ultracentrifuged at 100,000 ×g for 70 min. The pellet obtained was resuspended in phosphate-buffered saline (PBS) and subjected to a second ultracentrifugation at 100,000 ×g for 70 min. The precipitate obtained represented the sEVs, which were resuspended in PBS (HyClone; USA) for nanoparticle tracking analysis using NanoSight NS300 (Malvern Instruments Ltd.; UK) and transmission electron microscopy (TEM) imaging (FEI; Hillsboro, USA) and in radioimmunoprecipitation assay (RIPA) buffer (Beyotime; China) for western blotting.
TEM imaging
Purified sEVs were fixed in 1% glutaraldehyde for 2 h and placed on a 200-mesh Formvar-carbon-coated electron microscopy grid for 5 min at room temperature. The samples were stained with phosphotungstic acid for 5 min and dried for 10 min. The sEVs were then visualized using an FEI Tecnai F20 S TEM at 200 kV.
Animal experiment
An animal model of I/R was established as previously described. 30 Eight-weeks-old C57BL/6 mice were obtained from Shanghai Jie Si Jie Laboratory Animal Co., Ltd. (Shanghai, China), anesthetized by isoflurane inhalation (1.5%–2%), and ventilated with 95% oxygen (O2) and 5% carbon dioxide (CO2) using a small-animal ventilator. The mice were randomly assigned to different groups. The body temperature of the mice was maintained at 37°C using a warming pad. The mice were intravenously injected with 100 μL of VEC-derived sEVs (approximately 7 × 1012 particles calculated using NanoSight NS300, representing the concentration with the highest treatment efficiency and lowest adverse reactions) or an equal volume of PBS 4 h prior to myocardial I/R injury. An oblique incision was made in the chest of the mice to expose the left anterior descending coronary artery. The artery was ligated 1 mm below the tip of the left atrial appendage with a 6-0 silk suture to block blood flow for 45 min, followed by reperfusion for 24 h. A small-animal electrocardiogram monitoring system was used to detect ST-segment elevation. The mice were sacrificed by cervical dislocation. The heart was frozen at −20°C for 30 min and sliced at 2 mm/slice. Transferase dUTP nick-end labeling (TUNEL) staining and western blotting were performed to assess cardiomyocyte apoptosis. Cardiac function was evaluated using small-animal echocardiography. The experimental protocol was approved by the Ethics Committee of Shanghai General Hospital (2021AW035). All procedures were performed in accordance with the Declaration of Helsinki 1975, as revised in 2024.
Measurement of enzyme activity
After 24 h of reperfusion, 10 μL of serum samples were collected from the mice to measure enzymatic activities of lactate dehydrogenase (LDH), creatine kinase (CK), and CK-MB using commercial kits ab102526 (Abcam; UK), ab155901 (Abcam; UK), and ab193696 (Abcam; UK), respectively.
Echocardiography
The myocardium of the mice was assessed using M-mode echocardiography 24 h after reperfusion. A 14-Hz ultrasound probe (Hewlett-Packard Sonos 5500; USA) was used to monitor cardiac structure and function. The mice were anesthetized with diethyl ether and ventilated with 95% O2 and 5% CO2 using a small-animal ventilator.
Isolation and culture of cardiomyocytes
Primary cardiomyocytes were isolated from neonatal mice (1–3 days after birth) as previously described. 31 The hearts were excised into 1–3 mm pieces, digested with 0.2 mg/mL collagenase II (Worthington, USA), and incubated at 37°C for 5 min. The supernatant was then collected after three repetitions. The supernatant was spread on 10-cm sterile culture dishes (Corning; USA), and the cardiomyocytes and fibroblasts were separated after 1.5 h. Primary cardiomyocytes were cultured in DMEM containing 10% FBS at 37°C with 5% CO2. For the I/R cell model, the cardiomyocytes were cultured under 95% nitrogen (N2) and 5% CO2. The cells were reoxygenated 4 h later for another 4 h at 37°C with 5% CO2.
Cellular uptake of sEVs in vitro
The sEV uptake was measured using fluorescence experiments. Cardiomyocytes were cultured with VEC-derived sEVs and treated with PKH26 for 0.5 and 2 h. The PKH26 red fluorescence labeling kit (Sigma; USA) was used to investigate the cellular uptake of sEVs following the manufacturer’s recommendations. A 1 mM stock solution was prepared by dissolving 1 mg PKH26 in 1 mL dimethyl sulfoxide. This stock solution was diluted with serum-free cell culture medium at a ratio of 1:10 to obtain a 100 μM working solution. The dye working solution was added to the sEVs (50 μL of dye working solution is recommended for 10–200 μg of exosomal protein), and the mixture was vortexed for 1 min and incubated for 10 min. Subsequently, 10 mL of 1× PBS was mixed thoroughly with the incubated sEV–dye complex. The sEVs were extracted again using the sEV extraction method to remove excess dye, and 200 μL of 1× PBS was used to resuspend the precipitate, yielding fluorescently labeled sEVs. Cardiomyocytes were seeded in a 24-well plate and cultured overnight in serum-free medium. The following day, PKH26-labeled sEVs (20 μL) were added to the cells and cultured for the required duration. After washing with PBS, the cardiomyocytes were fixed in 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 (Sigma, USA) at 25°C for 20 min, and blocked with 3% bovine serum albumin for 1 h. Finally, 4′,6-diamidino-2-phenylindole (DAPI) was added and incubated for 10 min. Images were captured using a laser-scanning confocal microscope (Leica Microsystems; Germany).
RNA extraction and relative quantitative real-time polymerase chain reaction (RT-PCR)
VECs and their derived sEVs were used to assemble miRNA libraries. Following the manufacturer’s instructions, total RNA from cells was extracted using TRIzol reagent and RNA from VEC-derived sEVs using TRIzol LS (Thermo Fisher; USA). Ultraviolet spectrophotometry was used to determine the quality and purity of RNA at 260 nm (NanoDrop 2000, Thermo Scientific; USA). The extracted RNA was sent to GENEWIZ Bio (Suzhou, China) for miRNA transcriptome sequencing. Complementary DNA (cDNA) was synthesized from 1 μg of total RNA using a PrimeScript RT reagent kit (TaKaRa; Japan). Relative quantitative RT-PCR was performed using the iTaq Universal SYBR Green Supermix kit (TaKaRa; Japan). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and U6 were used as endogenous controls for mRNAs and miRNAs, respectively. The primer sequences used in this study were as follows: GAPDH forward primer 5′-
Fluorescence in situ hybridization (FISH)
A Cy5-labeled probe (Servicebio; China) targeting smad7 mRNA and a carboxyfluorescein (FAM)-labeled probe (Servicebio; China) targeting miR410-3p were used for FISH. Cardiomyocytes were cultured to 80%–90% confluence. After prehybridization, the cardiomyocytes were hybridized with Cy5- and FAM-labeled probes in hybridization buffer at 60°C overnight, and the cellular nuclei were stained with DAPI (Beyotime Biotechnology; China). All images were obtained using a microscope (Leica Microsystems; Germany).
Dual-luciferase reporting assay
The wild-type or mutant type of the 3′ untranslated region (3′UTR) of SMAD7 mRNA was inserted upstream of the luciferase 2 gene in the pmirGLO vector (Promega; USA). The vector was cotransfected into 293T cells with miR410-3p using Lipofectamine 3000 (Thermo Scientific; USA). After 48 h of transfection, the 293T cells were lysed, and luciferase activity was measured using the Dual-Luciferase Assay Kit (Promega; USA) and a plate reader (Thermo Scientific; USA). Renilla luciferase was used as the normalization reference.
Western blotting
Cells or tissues were lysed in RIPA buffer containing a protease inhibitor cocktail and centrifuged at 10,000 ×g for 30 min. Proteins were quantified using a bicinchoninic acid (BCA) kit (Beyotime Biotechnology; China). An equal amount (20 μg) of protein was loaded into each well and separated by 12.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, followed by transfer onto a polyvinylidene fluoride (PVDF) membrane (Millipore; MA). After blocking for 2 h with 5% nonfat milk in 1× Tris-buffered saline (TBS) with 0.1% Tween® 20 detergent, the PVDF membranes were incubated overnight at 4°C with primary antibodies, including anticleaved Poly (ADP-ribose) polymerase 1 (PARP1) (1:1000; Abcam; UK), anticleaved caspase-3 (1:1000; Abcam; UK), anti-SMAD7 (1:1000; Proteintech; USA), anti-beta-tubulin (1:5000; Abcam; UK), anti-GAPDH (1:5000; Cell Signaling Technology; USA), and anti-CD9/CD63/CD81/TSG101 (1:1000; Sangon Biotech; China). The PVDF membranes were washed thrice and subsequently incubated with antirabbit and antimouse secondary antibodies labeled with horseradish peroxidase (1:2000; Abcam; UK) for 2 h at room temperature. Immunoblotting was performed using the Gel Doc XR+ and ChemiDoc XRS+ Imaging Systems (Bio-Rad; USA). GAPDH served as the normal endogenous reference protein.
TUNEL
Tissue and cell apoptosis were measured using a TUNEL assay kit (Roche; Switzerland). Samples were dehydrated in a xylene–ethanol gradient and permeabilized with 1% proteinase K for 30 min. After rinsing with TBS, the samples were quenched with 3% hydrogen peroxide (H2O2) for 5 min and equilibrated with terminal deoxynucleotidyl transferase (TdT) equilibration buffer for 30 min, followed by incubation with TdT labeling reaction mix for 1.5 h at 37°C. The samples were then incubated with the stop solution for 5 min. After rinsing and blocking at room temperature, the samples were immersed in a 1× conjugate for 30 min. The samples were subsequently incubated with 3,3′-diaminobenzidine solution for 15 min. After washing three times, they were counterstained with methyl green for 3 min. Finally, the samples were dehydrated and imaged under a fluorescence microscope (Leica Microsystems; Germany).
Flow cytometry
Cardiomyocyte apoptosis was evaluated using flow cytometry. Cardiomyocytes treated with I/R and VEC-derived sEVs were detached with trypsin without ethylenediaminetetraacetic acid and washed three times with PBS. The cells were incubated with 5 μL fluorescein isothiocyanate and 5 μL propidium iodide for 20 min at room temperature and analyzed using flow cytometry (BD Accuri C6, BD Biosciences; USA).
RNA immunoprecipitation assay
Cardiomyocytes were transfected for 24 h with miR410-3p mimics or an over-smad7 plasmid using Lipo3000 (Thermo Scientific; USA) and then lysed with RNA immunoprecipitation lysis buffer containing RNase and protease inhibitors, following the manufacturer’s instructions (Millipore; USA). After treatment with DNase I at 37°C for 10 min and centrifugation at 12,000 ×g for 30 min, the lysate was incubated overnight with an Argonaute-2 (AGO2) antibody (Abcam, UK; 1:50; ab186733) or an immunoglobulin G (IgG) antibody (Abcam, UK; 1:50; ab172730) at 4°C. RNA was then purified and reverse-transcribed into cDNA for RT-PCR analysis.
Statistical analysis
All data were presented as mean ± SD from at least three independent experiments. Statistical significance was determined using Student’s t-test for two groups or analysis of variance followed by Tukey’s test for more than two groups. Data were analyzed using GraphPad Prism 8.0 (GraphPad Software, USA). A p value <0.05 was considered statistically significant.
Results
Isolation and characterization of VEC-derived sEVs
This study aimed to elucidate the effect of VEC-derived sEVs on I/R-induced cardiac injury. VECs were cultured for 72 h in a FBS-free DMEM medium. The supernatant (200 mL) was collected, and sEVs were isolated using ultracentrifugation (Figure 1(a)). The morphology of multivesicular bodies (MVBs) and sEVs was assessed using TEM, which revealed round vesicles with a double-layered membrane structure (Figure 1(b) and (c)). The size distribution of the extracellular particles was quantified using a nanoparticle tracking system, revealing diameters ranging from 50 to 200 nm, with a predominant peak diameter of 120 nm (Figure 1(d)). Western blot analysis confirmed the extracellular vesicle (EV) nature of these particles, demonstrating positive reactivity for EV markers, including CD9, CD63, CD81, and TSG101 (Figure 1(e)). These findings provided conclusive evidence that the extracellular particles released by VECs in this study exhibited characteristics consistent with sEVs.

Isolation and identification of VEC-derived sEVs. (a) Isolation scheme of VEC-derived sEVs. (b) Morphology of sEVs in MVB in VECs was imaged using TEM. Scale bar: 500 nm. Data represent one of three independent experiments. (c) Ultrastructure of VEC-derived sEVs was analyzed using TEM. Scale bar: 100 nm. Data represent one of three independent experiments. (d) NanoSight was used to measure the diameter range of VEC-derived sEVs. Data represent one of three independent experiments. (e) Expression of exosomal protein markers (CD9, CD81, CD63, and TSG101) in sEVs and VECs was measured using western blotting. Data represent one of three independent experiments. MVB: multivesicular bodies; sEVs: small extracellular vesicles; TEM: transmission electron microscopy; VEC: vascular endothelial cell; CD: cluster of differentiation; TSG101: tumor susceptibility gene 101.
Administration of VEC-derived sEVs alleviated I/R-induced myocardial injury in vivo
To investigate the effect of VEC-derived sEVs on I/R-induced myocardial injury, a mouse I/R model was established and evaluated using an electrocardiogram showing ST-segment elevation. The mice were then administered VEC-derived sEVs via tail vein injection (Figure 2(a)). The enzymatic activities of LDH, CK, and CK-MB in the serum of mice with I/R were quantified using commercial kits to assess the extent of injury. The LDH, CK, and CK-MB levels were elevated in the I/R group compared with those in the sham group, indicating I/R-induced myocardial injury. Conversely, mice with I/R who were treated with VEC-derived sEVs exhibited lower enzymatic activities, suggesting that the sEVs attenuated I/R-induced myocardial injury (Figure 2(b) to (d)). Furthermore, color Doppler echocardiography revealed improved cardiac function parameters, including left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic diameter (LVESD), left ventricular fractional shortening (LVFS), and left ventricular ejection fraction (LVEF), in mice treated with VEC-derived sEVs (Figure 2(e) to (i)). Western blotting confirmed that VEC-derived sEVs downregulated cleaved PARP1 and cleaved caspase-3 expression (Figure 2(j)). The terminal deoxynucleotidyl TUNEL assay showed that VEC-derived sEVs reduced cardiomyocyte apoptosis (Figure 2(k)). Overall, VEC-derived sEVs demonstrated a marked inhibitory effect on I/R-induced cardiomyocyte apoptosis.
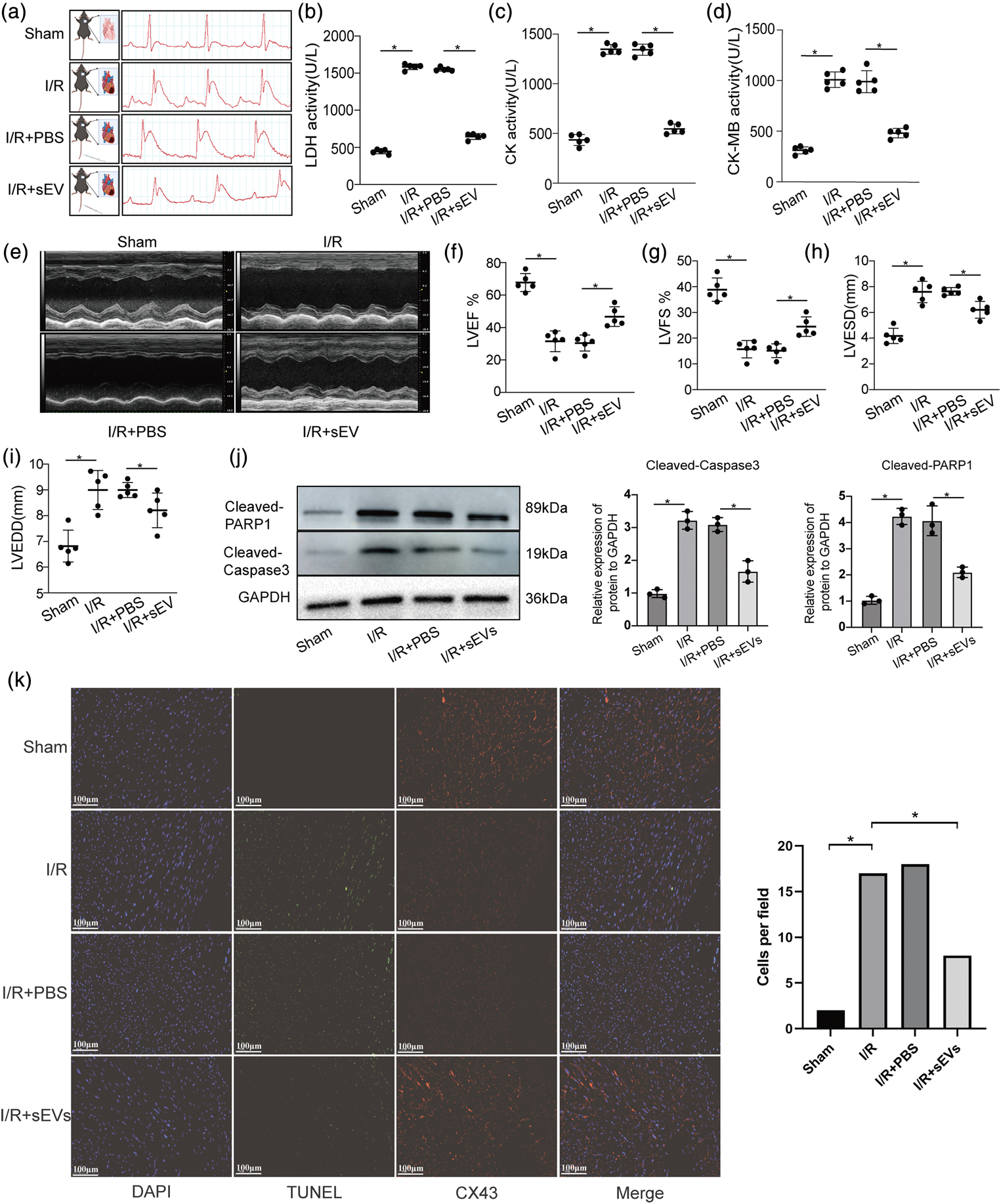
VEC-derived sEVs alleviated I/R-induced myocardial injury in vivo. (a) Diagram of the I/R-induced myocardial injury mouse model with and without pretreatment with VEC-derived sEVs (left). The I/R-induced myocardial injury model was confirmed using an electrocardiogram showing ST-segment elevation (right). Data represent one of three independent experiments. (b–d) Activities of LDH, CK, and CK-MB in mouse serum from the four groups were measured using commercial kits. Data are presented as mean ± SD (n = 5 per group, *p < 0.05). (e) Cardiac function in the four mouse groups was assessed using transthoracic echocardiography. Data represent one of three independent experiments. (f–i) Main parameters analyzed using transthoracic echocardiography: (F) LVEF; (G) LVFS; (H) LVESD; and (I) LVEDD. Data are presented as mean ± SD (n = 5 per group, *p < 0.05). (j) Apoptosis protein markers (cleaved PARP1 and caspase-3) were Continued.measured using western blotting and quantitative analysis. Data represent one of three independent experiments. (k) Cardiomyocyte apoptosis was determined using the TUNEL assay. Blue indicates DAPI, green indicates TUNEL, and red indicates CX43, expressed mainly in cardiomyocytes. Scale bar: 100 µm. Data represent one of three independent experiments. Quantitative data are presented as mean ± SD (*p < 0.05). CK: creatine kinase; DAPI: 4′,6-diamidino-2-phenylindole; I/R: ischemia–reperfusion; LDH: lactate dehydrogenase; LVEF: left ventricular ejection fraction; LVFS: left ventricular fractional shortening; LVESD: left ventricular fractional shortening; LVEDD: left ventricular end-diastolic diameter; sEVs: small extracellular vesicles; TUNEL: transferase dUTP nick-end labeling; VEC: vascular endothelial cell; PARP1: Poly (ADP-ribose) polymerase 1.
VEC-derived sEVs suppressed I/R-induced cardiomyocyte apoptosis in vitro
Primary cardiomyocytes were used to investigate the role of VEC-derived sEVs in I/R-induced cardiac injury in vitro. Endocytosis experiments were performed to measure the uptake of VEC-derived sEVs by cardiomyocytes (Figure 3(a)). Fluorescence results demonstrated efficient endocytosis of sEVs by cardiomyocytes (Figure 3(b)). Flow cytometry analysis further confirmed that VEC-derived sEVs inhibited I/R-induced cardiomyocyte apoptosis (Figure 3(c)). Western blotting demonstrated that VEC-derived sEVs reduced the expression of cleaved PARP1 and cleaved caspase-3 in cardiomyocytes with I/R treatment (Figure 3(d)). These findings suggest that VEC-derived sEVs attenuate I/R-induced cardiomyocyte apoptosis in vitro.

VEC-derived sEVs decreased I/R-induced cardiomyocyte apoptosis in vitro. (a) Diagram of PKH26-labeled VEC-derived sEV preparation and endocytosis experiments. (b) Uptake of PKH26-labeled VEC-derived sEVs by cardiomyocytes was examined after specific incubation times (30 min and 2 h). Blue indicates DAPI; red indicates PKH26, which is associated with sEVs. Scale bar: 20 μm. Data represent one of three independent experiments. (c) I/R-induced cardiomyocyte apoptosis with or without treatment with VEC-derived sEVs was determined using flow cytometry and quantitative analysis. Data represent one of three independent experiments. (d) Apoptosis protein markers (cleaved PARP1 and caspase-3) were measured using western blotting and quantitative analysis. Data represent one of three independent experiments. DAPI: 4′,6-diamidino-2-phenylindole; I/R: ischemia–reperfusion; sEVs: small extracellular vesicles; VEC: vascular endothelial cell; PARP1: Poly (ADP-ribose) polymerase 1.
VEC-derived sEVs miR410-3p inhibited I/R-induced cardiomyocyte apoptosis
The miRNAs are key mediators of sEVs involved in various pathological processes.32,33 We explored whether miRNAs in VEC-derived sEVs were involved in I/R-induced myocardial injury by analyzing the expression profiles of miRNAs in VECs and VEC-derived sEVs using next-generation sequencing technology. The Venn diagram in Figure 4(a) shows that 237 known miRNAs were expressed in VECs and VEC-derived sEVs. A total of 59 known miRNAs were expressed only in VECs, whereas 23 were expressed only in sEVs. Additionally, miR410-3p was strongly expressed in sEVs (Figure 4(b)). Having identified miR410-3p as a potential candidate associated with the pathological process of apoptosis,34–36 we investigated its role in I/R-induced myocardial injury.

VEC-derived sEV miR-410-3p inhibited I/R-induced cardiomyocyte apoptosis. (a) Venn diagram of known miRNAs in VECs and VEC-derived sEVs. (b) Ten most abundantly expressed miRNAs in VECs and VEC-derived sEVs. (c) Diagram of the VEC (top layer) and cardiomyocyte (bottom layer) coculture model. Red dots represent sEVs; blue dots represent GW4869. (d) Expression of miR-410-3p in cardiomyocytes cocultured with VECs pretreated with or without GW4869. Data are presented as mean ± SD (n = 3 per group, *p < 0.05). Data represent one of three independent experiments. (e) I/R-induced cardiomyocyte apoptosis with or without treatment with VEC-derived sEVs, miR410-3p mimics, and miR410-3p inhibitors was determined using flow cytometry and quantitative analysis. Data represent one of three independent experiments. (f) I/R-induced cardiomyocyte apoptosis protein markers (cleaved PARP1 and caspase-3) with or without treatment with VEC-derived sEVs, miR410-3p mimics, and miR410-3p inhibitors were measured using western blotting and quantitative analysis. Data represent one of three independent experiments.
Cocultivation experiments, using a Transwell system with a 0.4 μm Transwell insert pore size, were performed to determine whether miR410-3p in VEC-derived sEVs was involved in I/R-induced cardiomyocyte apoptosis (Figure 4(c)). VEC-derived sEVs significantly increased miR410-3p expression in cardiomyocytes, whereas pretreatment with GW4869, a sphingomyelinase inhibitor that inhibits sEV biogenesis/release, reduced this effect (Figure 4(d)). Manipulation of miR410-3p levels in cardiomyocytes using mimics or inhibitors demonstrated that its overexpression enhanced the antiapoptotic function of VEC-derived sEVs, whereas its inhibition counteracted this effect (Figure 4(e)). Western blotting showed that miR410-3p overexpression reduced the expression of cleaved PARP1 and cleaved caspase-3 (Figure 4(f)). These findings indicate that miR410-3p is a key mediator of VEC-derived sEVs in protecting against cardiomyocyte apoptosis.
miR410-3p regulated cardiomyocytes apoptosis by targeting SMAD7
The miRNAs regulate cellular processes primarily by modulating protein expression through interactions with target mRNAs. 37 To identify the downstream target of miR410-3p involved in regulating cardiomyocyte apoptosis in I/R-induced cardiac injury, bioinformatics tools such as miRanda and TargetScan were used to predict target mRNAs. Based on the comprehensive score, smad7 was identified as a potential target mRNA. FISH analysis demonstrated that miR410-3p and smad7 mRNA were colocalized in the cardiomyocyte cytoplasm (Figure 5(a)). A conserved binding site for miR410-3p was identified in the 3′UTR of SMAD7 mRNA (Figure 5(b)). The miRNAs bind to AGO2 and target mRNAs to form an RNA-induced silencing complex, thereby regulating protein expression. We performed an AGO2-based RNA immunoprecipitation assay to examine the enrichment between miR410-3p and the smad7 gene. The results showed that smad7 overexpression increased miR410-3p enrichment in AGO2-immunoprecipitated transcripts, and miR410-3p mimics enhanced smad7 enrichment (Figure 5(c) and (d)). To verify whether miR410-3p directly interacts with the 3′UTR of smad7, we constructed luciferase reporter plasmids, including the mutant SMAD7 3′UTR (pGL3-SMAD7mut‑3′UTR) and the wild-type SMAD7 3′UTR (pGL3-SMAD7WT‑3′UTR) (Figure 5(b)). Following cotransfection with miR-410-3p mimics, a 30% reduction in luciferase activity in the pGL3-SMAD7WT‑3′UTR group was observed compared with that in the pGL3-SMAD7mut‑3′UTR group (Figure 5(e)). Real-time quantitative polymerase chain reaction (RT-qPCR) and western blotting results demonstrated that VEC-derived sEVs significantly decreased smad7 mRNA and protein expressions at a concentration of 20 μL/mL (Figure 5(f) and (g)). Furthermore, the downregulation of sEV-derived miR410-3p inhibited the effect of VEC-derived sEVs on SMAD7 protein expression (Figure 5(h)). Meanwhile, miR410-3p significantly reduced smad7 mRNA and SMAD7 protein expressions (Figure 5(i) and (j)).

SMAD7 mediated miR410-3p-associated cardiomyocyte apoptosis. (a) Colocalization of smad7 and miR410-3p in the cardiomyocyte cytoplasm was examined using FISH. Scale bar: 20 μm. Blue: DAPI; green: Cy5-miR410-3p; red: FAM-smad7. Data represent one of three independent experiments. (b) Schematic representation of the pmirGLO plasmids with smad7-WT and smad7-mut for the dual-luciferase reporter assay. (c, d) AGO2-RNA RIP in cardiomyocytes transfected with smad7 (C) or miR410-3p mimics (D). Levels of miR410-3p or smad7 mRNA were measured using quantitative PCR. RIP represents an RNA-binding protein IP assay; 10% of samples were loaded. Data are presented as mean ± SD (n = 3 per group, *p < 0.05). Data represent one of three independent experiments. (e) Luciferase activity was analyzed using quantitative PCR. Data are presented as mean ± SD (n = 3 per group, *p < 0.05). Data represent one of three independent experiments. (f, g) Expression of smad7 mRNA (F) and protein (G) in cardiomyocytes treated with a specific concentration of VEC-derived sEVs was measured using quantitative PCR and western blotting. Data are presented as mean ± SD (n = 3 per group, *p < 0.05). Data represent one of three independent experiments. (h) Expression of SMAD7 protein in cardiomyocytes treated with VEC-derived sEVs with or without si-miR410-3p was tested using western blotting and quantitative analysis. Data represent one of three independent experiments. (i, j) Expression of smad7 mRNA (I) and protein (J) in cardiomyocytes treated with miR410-3p mimics was measured using quantitative PCR and western blotting. Data are presented as mean ± SD (n = 3 per group, *p < 0.05). Data represent one of three independent experiments. (k, l) I/R-induced cardiomyocyte apoptosis protein markers (cleaved PARP1 and caspase-3) Continued.with or without treatment with miR410-3p mimics and SMAD7 overexpression were measured using western blotting (K) and quantitative analysis (L). Data represent one of three independent experiments. (m) I/R-induced cardiomyocyte apoptosis with or without treatment with miR410-3p mimics and SMAD7 overexpression was measured using flow cytometry. Data represent one of three independent experiments. DAPI: 4′,6-diamidino-2-phenylindole; FISH: fluorescence in situ hybridization; I/R: ischemia–reperfusion; PCR: polymerase chain reaction; sEVs: small extracellular vesicles; VEC: vascular endothelial cell; SMAD7: SMAD family member 7; mRNA: messenger ribonucleic acid; PARP1: Poly (ADP-ribose) polymerase 1; FAM: carboxyfluorescein; WT: wild-type; mut: mutant; AGO2: Argonaute2; RIP: RNA immunoprecipitation; IP: immunoprecipitation.
Moreover, we constructed SMAD7 overexpression plasmids to investigate whether SMAD7 is associated with the function of miR410-3p in I/R-induced cardiomyocyte apoptosis. This was achieved by cotransfecting miR410-3p mimics with SMAD7 overexpression plasmids into cardiomyocytes. Western blotting showed that miR410-3p reduced the expression of cleaved PARP1 and cleaved caspase-3. However, SMAD7 overexpression inhibited the effect of miR-410-3p (Figure 5(k) and (l)). Flow cytometry analysis demonstrated that SMAD7 overexpression enhanced miR410-3p-related cardiomyocyte apoptosis (Figure 5(m)). These findings suggest that miR410-3p modulates cardiomyocyte apoptosis by regulating SMAD7 protein expression.
Discussion
The primary goal of myocardial infarction treatment is to restore coronary perfusion to preserve cardiac tissue. 1 However, reperfusion can result in severe and irreversible myocardial injury. 38 As VECs are the predominant cell type in myocardial tissue, endothelial cell (EC)–derived sEVs are abundant and actively contribute to various pathological processes, including inflammation, apoptosis, and fibrosis. 39 The sEV-encapsulated miRNAs have attracted considerable attention because of their central role in mediating sEV functions in biological activities. 40 Herein, we demonstrated that VEC-derived sEVs inhibit I/R-induced myocardial injury. Furthermore, we elucidated the intricate molecular mechanisms underlying this protective effect. Our findings demonstrated that VEC-derived sEVs suppressed cardiomyocyte apoptosis through the action of miR410-3p, which suppresses SMAD7 expression.
Recent studies have reported various effects of sEVs on I/R-induced myocardial injury. Yadid et al. 41 attributed the protective effects of endothelial EVs to the presence of protective proteins, including antioxidants, enzymes, adenosine monophosphate–activated protein kinase–related proteins, heat shock proteins, and proteins involved in cellular redox homeostasis or the unfolded protein response pathway. Su et al. 42 further corroborated the protective role of VEC-derived sEVs by emphasizing the involvement of long noncoding RNA (lncRNA). Livkisa et al. 43 confirmed that sEVs derived from human platelets strongly protect against cardiac I/R injury. In our study, administration of VEC-derived sEVs via tail vein injection successfully attenuated cardiac injury in a mouse I/R model. Furthermore, previous studies have shown that VEC-derived sEV miRNA24-3p attenuates myocardial I/R injury by preventing Ly6Chigh monocyte recruitment. 44 Our findings provide new insights and demonstrate the versatile roles of EC-derived sEVs in cardiac diseases.
Subsequent analysis of miRNA content in VEC-derived sEVs revealed an abundance of miR410-3p. The miR410-3p promotes cardiac hypertrophy and is involved in cellular apoptosis in various cardiac diseases.35,36,45 Therefore, we explored whether miR410-3p is a principal mediator of VEC-derived sEVs during I/R. In vitro experiments demonstrated that the upregulation of miR410-3p together with VEC-derived sEVs effectively inhibited cardiomyocyte apoptosis. Conversely, miR410-3p inhibition weakened the protective effect of VEC-derived sEVs, highlighting the association between miR410-3p and VEC-derived sEVs as a protective factor in cardiac I/R processes.
Additionally, the molecular mechanisms by which miR410-3p attenuates I/R-induced cardiac injury were elucidated. Prediction software identified SMAD7 as a potential target of miR410-3p. Experimental validation confirmed that miR410-3p binds to the 3′UTR of SMAD7 mRNA and its overexpression reduced SMAD7 mRNA and protein levels in cardiomyocytes. Furthermore, the protective effect of miR410-3p on I/R-induced cardiomyocyte apoptosis was neutralized by SMAD7 overexpression, which inhibited the antiapoptotic protein nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). 46 These findings indicate that miR410-3p protects against I/R-induced cardiomyocyte apoptosis by downregulating SMAD7 expression.
SMAD7 belongs to the SMAD protein family, which plays a vital role in the classical Transforming growth factor-beta (TGF-β) pathway (SMAD-dependent TGF-β pathway). In the classical TGF-β pathway, SMAD7 inhibits signaling by competitively binding to the TGF-β1 receptor and promoting its degradation. 47 However, Edlund et al. 48 reported that SMAD7 enhances the nonclassical TGF-β signaling pathway (non-SMAD-dependent TGF-β signaling pathway) by acting as a scaffold protein to activate TGF-β-activated kinase 1 (TAK1), thereby activating p38 mitogen-activated protein kinase (p38 MAPK). Therefore, SMAD7 exerts contrasting effects on the TGF-β signaling pathway through these two mechanisms.
Another study reported that miR-410-3p targets SMAD7 mRNA, exacerbating experimental drug (AngII)–induced cardiac hypertrophy. 36 The miR-410-3p/SMAD7 axis appears to play different roles in cardiac diseases. SMAD7 inhibits the classical TGF-β signaling pathway and alleviates cardiac fibrosis and cardiac hypertrophy mediated by the SMAD-dependent TGF-β pathway. 49 However, in the early stages of myocardial I/R injury, oxidative stress and apoptosis are the primary pathological processes, making the non-SMAD-dependent TGF-β signaling pathway more relevant to I/R injury–mediated apoptosis. 50 Therefore, the miR-410-3p/SMAD7 axis may exert different effects across different cardiac diseases. Moreover, different cardiac diseases involve different predominant cell types (such as fibroblasts and cardiomyocytes), which may also explain the varying effects of the miR-410-3p/SMAD7 axis in different cardiac conditions.
However, this study has certain limitations. For instance, when examining the impact of the interaction between miR-410-3p and SMAD7 on myocardial I/R injury, we did not establish miR-410-3p knockout mice or SMAD7 knockout mice models. Additionally, several studies have reported that the biological functions of miRNAs are regulated by upstream lncRNAs; however, our study focused only on the downstream effects of miR-410-3p and did not investigate its upstream regulatory mechanisms.
Conclusion
Our study confirms the crucial roles of VEC-derived sEVs in I/R-induced cardiac injury and delineates the underlying mechanism involving miR410-3p-mediated inhibition of SMAD7 (Figure 6). These findings provide a robust foundation for future in-depth investigations into the nuanced influence of VEC-derived sEVs on the cardiac I/R process.
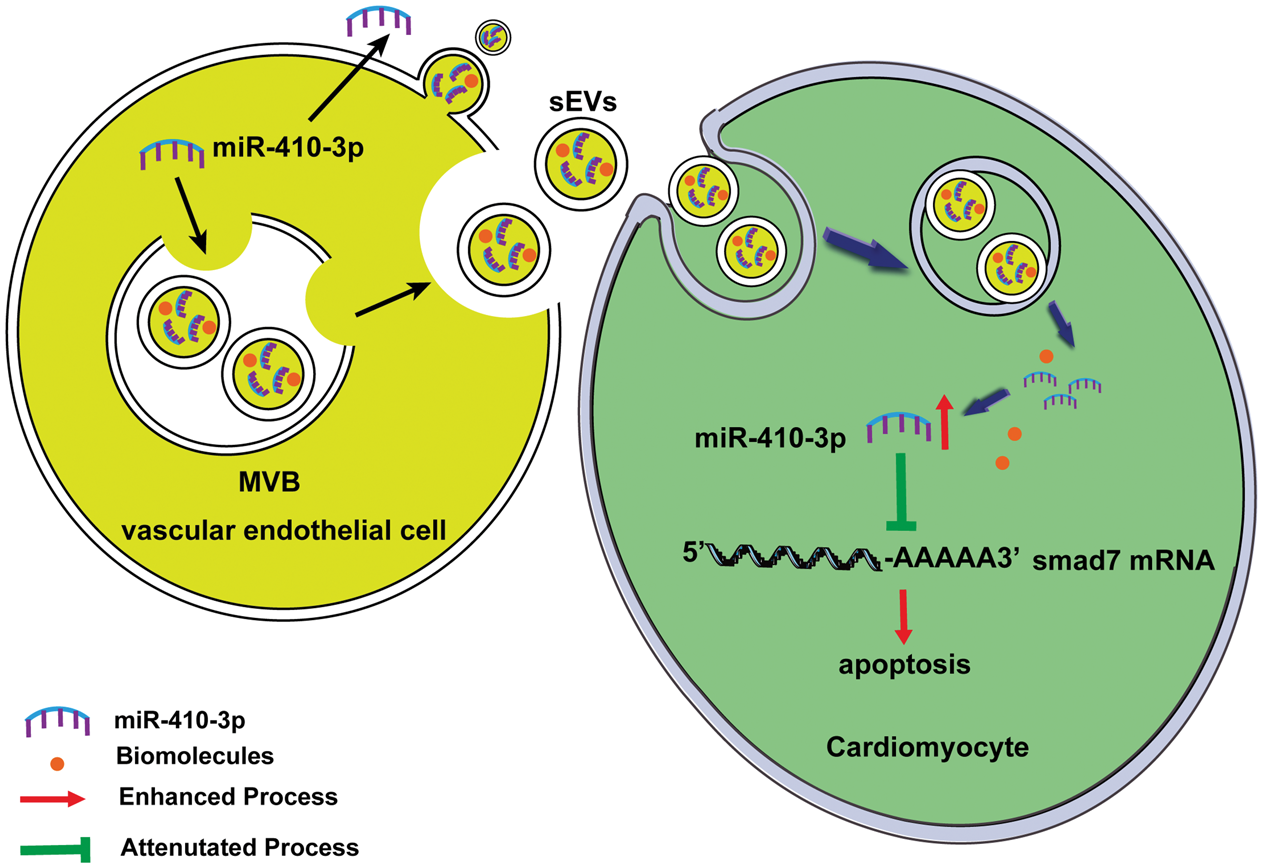
miR410-3p inhibits cardiomyocyte apoptosis via SMAD7 regulation. SMAD7: SMAD family member 7.
Footnotes
Acknowledgment
Not applicable.
Authors’ contributions
FW and WL designed the study. FW and WL supervised the project. XL, YG, and JL performed all experiments. XX, HC, DW, and HN analyzed the data. WL wrote the manuscript. All authors read and approved the final manuscript.
Availability of data and materials
Not applicable.
Declaration of conflicting interests
The authors declare that they have no competing interests.
Funding
This work was supported by the National Key Research and Development Program of China (No. 2023YFB3208200), the Science and Technology Tackling (Medical and Health Field) Project of the Shanghai Songjiang District Science and Technology Commission (No. 22SJKGGG15), and the National Natural Science Foundation of China (No. 82070334 and 82570371).