
Abstract
Objective
In the pathogenesis of the post-injury period following nonpenetrating traumatic brain injuries (TBIs; post-TBIs), pathological metabolic changes in the oxidative status are important, as a complete understanding of their interactions can explain the concept of how oxidative metabolic–related differences develop, thereby supporting effective treatment.
Methods
This case–control study included 63 patients (mean age ± SD, 36.19 ± 11.4 years) in the post-TBI group and 32 healthy controls. Participants were tested for creatine kinase, adenosine triphosphate, and adenosine diphosphate levels using enzyme-linked immunosorbent assays and for pyruvate and lactate levels using a spectrophotometric method according to standard manufacturer protocols.
Results
This study found abnormally decreased adenosine triphosphate and adenosine diphosphate levels in the general nonpenetrating post-TBI group versus controls. The median total adenosine triphosphate levels were 600.4 ± 46.96 in the general clinical group and 745.7 ± 49.83 mkmol/L in controls (p < 0.0001, t = 13.69, 95% confidence interval: 124.0 to 166.5). The median total adenosine diphosphate levels were 247.6 ± 33.09 in the general clinical group and 273.9 ±31.98 mkmol/L in controls (p = 0.0004, t = 3.734, 95% confidence interval: 12.20 to 40.26). Compared with controls, elevated creatine kinase levels were found in the general clinical group (p < 0.0001, t = 7.030, 95% confidence interval: −39.78 to −22.19). The median total creatine kinase (mean ± SD) levels were 129.6 ± 21.37 in the general clinical group and 98.61 ± 19.74 IU/L in controls. Higher creatine kinase (p < 0.0001, t = 4.779, 95% confidence interval: 13.16 to 32.23) and lower adenosine triphosphate levels (p < 0.001, t = 4.997, 95% confidence interval: −70.74 to −30.31) were found to be more pronounced after brain contusion compared with those following mild TBIs.
Conclusions
Nonpenetrating post-TBIs showed higher creatine kinase levels associated with lower adenosine triphosphate–adenosine diphosphate levels, indicating oxidative dyshomeostasis at 12-month post-injury follow-ups, which could play a pathogenetic role in post-TBIs progression.
Keywords
Introduction
Mild traumatic brain injuries (TBIs) and cerebral contusions are the most common forms of brain trauma, with an annual incidence estimated between 100 and 600 clinical cases per 100,000 people across different countries. They result in persistent neurological and/or psychiatric symptoms and cognitive impairment.1,2 TBIs are a significant public and economic burden and are among the leading causes of death and disability in young adults worldwide.1,3 They are recognized as a complex morpho-functional state involving primary and secondary injury. Primary brain injury predominantly comprises the initial tear or hemorrhage, whereas secondary TBI exacerbates the initial injury through cascades of various oxidative processes and multiple pathological reactions triggered by the initial neurotrauma.4–7 Today, there is no doubt that various oxidative processes play an important role in the pathogenesis of post-traumatic encephalopathy, leading to the development of different pathological changes. However, the metabolic pathways underlying oxidative status and their mechanisms remain poorly understood. These observations underscore the need for objective tools to identify at-risk patients and to monitor disease-related symptoms and disease progression.
There are numerous experimental and clinical studies reporting the baseline pathological pathways of oxidative dyshomeostasis associated with the progression of secondary consequences of brain injury. These include various changes, among which the most well-investigated are blood–brain barrier breakdown, astrocytic swelling, cerebrovascular oxidative stress,8–13 neurotransmitter reuptake following reactive astrogliosis, neurovascular autoregulatory dysfunction,13–15 glial injury and dysfunction, excitotoxicity, and aberrant ionic dyshomeostasis in neurons as well as progressive white matter deterioration5,16–18 and gaseous factors (i.e. nitric oxide), which are important for regulating inflammation and systemic homeostasis responses in injuries. 19 Brain injuries during the initial and post-traumatic periods lead to energy failure, severe oxidative status changes, and mitochondrial dysfunction, followed by increased neurotransmitter extrication. These events increase intracellular calcium, which trigger additional pathologic cascades, including oxidative stress, production of reactive oxygen species, and activation of nitric oxide pathway, which results in the generation of reactive nitrogen species.16,20,21
Metabolic pathways underlying oxidative status changes and their pathogenetic mechanisms during the post-injury period following TBIs (post-TBIs) have not been fully discussed. Several studies suggest that TBI itself exacerbates rapid reactive oxygen production and oxidative damage to neuronal components, leading to neuronal dysfunction or cell death.22–24
Attempts to identify the exact and complete mechanisms underlying the long-term consequences of TBIs remain incomplete. Adenosine triphosphate (ATP) and adenosine diphosphate (ADP) are key compounds that provide energy to drive multiple biochemical processes in all cells. They participate in glycolysis, intracellular and extracellular signaling, neurotransmission, ribonucleic acid synthesis, and neuroamino acids activation in protein synthesis.21,25–28 They are essential because large amount of free energy is released during hydrolysis. This process provides energy for cells, in which oxidation energy is trapped by phosphoryl transfer compounds used to form ATP. There are several well-established observations of post-TBIs data demonstrating excessive inflammatory cytokine production and endogenous release of toxic compounds, which are typically intracellular molecules that have reached the extracellular space following cell damage, and all these processes must be supported by energy metabolism with ATP assessment.8,9,21,26
Failed brain energy metabolism can be among the important promoters of other secondary injury cascades that contribute to post-TBI development. Metabolic biochemical pathways involved in ATP/ADP metabolism are principal to understanding their role in the post-TBI state. Any disturbances in energy processes are crucial factors determining secondary changes in neuronal function. In this context, investigating energy-formation processes and metabolic enzymes in this patient cohort presents substantial interest.
The aim of this investigation was to assess changes in the oxidative status in the post-injury period following nonpenetrating TBIs (nonpenetrating post-TBIs) under baseline conditions during 12–14 months of post-injury follow-ups, representing the recovery period, and to determine whether these systemic biomarkers involved in oxidative metabolism are elevated in this patient cohort.
Materials and methods
Study design and participants
We conducted a retrospective observational study with cross-sectional and longitudinal components. Target enrollment included patients in nonpenetrating post-TBIs and age- and sex-matched healthy controls (HCs). The general clinical group consisted of patients (n = 63) in nonpenetrating post-TBIs (mean age ± SD, 36.19 ± 11.4 years) and 32 HCs (32.4 ± 5.02 years); all participants were tested for creatine kinase (CK), ATP, ADP, pyruvate, and lactate levels under the baseline conditions. Patients included in this investigation were recruited from State Institute of Neurology, Psychiatry, and Narcology of National Academy of Medical Sciences of Ukraine (INPN NAMSU). All patients received vascular therapy and nootropic drugs in the Department of Neurosurgery and Paroxysmal States, INPN NAMSU. Clinical and paraclinical details were deidentified to prevent patient identification. To ensure patient privacy, all collected data were anonymized, and any personally identifiable information was removed. To minimize pharmacologic confounding, baseline sampling was performed during clinical stability. Neurological disability was quantified by the Expanded Disability Status Scale (EDSS) conducted and scored by the independent certified rater (MP). Demographic and clinical characteristics and disease history (duration, computed tomography (CT) findings, magnetic resonance imaging (MRI) data, EDSS, and prior treatments) were abstracted from the database of INPN NAMSU hospital records.
Exclusion criteria included a history of any decompensated somatic pathology, other concomitant neurological pathology (except post-traumatic seizures), childhood brain trauma, penetrating TBI, psychiatric disorder or mental retardation, sepsis, autoimmune encephalitis, active infection, craniectomy, and inflammatory diseases present before sample collection and in their anamnesis. Patients receiving regular psychiatric and antiepileptic medications were excluded to rule out the potential effect of these medications on biochemical enzyme levels. Only patients with complete clinical and laboratory records were included. Additionally, patients with baseline cognitive impairment, pregnancy, diabetes mellitus with complications, intoxication at the time of enrollment, uncontrolled seizure disorder, use of anticoagulant drugs, and homelessness were excluded. Diagnosis was primarily based on clinical criteria and CT scans. An abnormal CT was defined as the presence of intracranial hematoma, subarachnoid hemorrhage, herniation, or compressed basal cistern. This retrospective cohort included adult trauma patients (age >18 years). All participants were evaluated for seizure frequency using diaries. HCs had no history of neurological or psychiatric diseases. All patients and HCs were sporadic and were born in Kharkiv, Ukraine.
TBI severity was classified according to the duration of post-traumatic amnesia, Glasgow Coma Scale, and duration of loss of consciousness. The diagnosis of TBI was established on a clinical basis, supported by neurosurgery consultations and CT and/or MRI findings. There is no single or unified system in the United States or European countries that tracks the occurrence of brain injuries across large populations. For reporting of sex and gender data in this study, the Sex and Gender Equity in Research guidelines (2016) were followed in accordance with best practices when considering sex and gender as variables and improving the generalizability of the findings.
Methods
Serum sampling for CK, ATP, and ADP determinations was performed using a 5.0-mL tube. Samples were allowed to clot for 30–35 min at room temperature and then centrifuged at 3000 ×g for 30 min. After centrifugation, serum was separated and frozen at −30°C to −80°C until analysis. CK, ATP, and ADP levels were measured via enzyme-linked immunosorbent assay (ELISA), according to standard manufacturer practices, using commercially available human ELISA and Dialab kits (Kharkiv National Medical University, Biochemistry Department).
Plasma sampling for pyruvate and lactate determination was performed using an 8.0-mL tube coated with sodium fluoride and ethylenediaminetetraacetic acid to prevent oxidation. Blood samples were processed as previously described in the protocol 29 and analyzed using the СФ46 spectrophotometer according to standard manufacturer instructions.
Controls, standards, and patient samples were analyzed in duplicate with a variation coefficient of <10%. All study protocols were validated by the local health ethics committee. Written and oral informed consent was obtained from each patient and control.
Procedures performed in this investigation involving human participants were in accordance with local health ethics committee guidelines and the Declaration of Helsinki of 1975, as revised in 2024. The local ethics committee approved the study protocol (Approval Number: 70-O), which was performed according to the ethical standards laid down in the Declaration of Helsinki.
Statistical analysis
All statistical analyses were performed using GraphPad Prism (version 10.5.0) on raw data. We described the baseline characteristics of our specific patient cohort and stratified subgroups according to the International Classification of Functioning, Disability, and Health. Group differences in demographics were tested via analysis of variance (ANOVA) or Kruskal–Wallis tests (age) and χ2 test (sex). All parametric tests were applied for normally distributed data, and nonparametric tests were used for non-normally distributed data. Mann–Whitney and Kruskal–Wallis tests were performed using Prism (v.10.5.0). Data were analyzed using two-way ANOVA, and a multiple-comparison test was applied to compare all values to controls. Repeated-measures ANOVA was also used to compare all variables. All tests were two-sided, and p values <0.05 were considered statistically significant. When analyzing serum metabolic enzyme levels as continuous variables, log transformation was applied to achieve normality. Data were presented as bar graphs with individual values and mean ± SEM.
Variables identified on statistical analysis
Comparisons were made across three groups: the general clinical group, which included patients in nonpenetrating post-TBIs at 12–14 months post-injury follow-ups; group 1, comprising patients in the post-injury period following mild TBIs; and group 2, comprising patients in the post-injury period following cerebral contusion. Mean, median, and proportion were used to present the distribution of the main characteristics of the study sample. To test differences in means, ANOVA was used, and χ² test and Fisher’s exact tests were applied to evaluate relationships between two categorical variables and differences in proportions.
Covariates
Data on patient demographics, baseline comorbidities, clinical characteristics, injury-related characteristics, and outcomes were collected and analyzed. A p value <0.05 was considered statistically significant and was applied to all experiments. The sample size was determined to achieve a statistical power of 0.8. Data were reported with a 95% confidence interval (CI). The dataset was created in Microsoft Excel, and statistical analysis was conducted using Analyze-it for Microsoft Excel version 4.92.4 and R software version 3.6.0.
The protocol defined the methods for literature critique/appraisal and used the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) tool to identify relevant content and methodology of the included studies. For preclinical animal studies, Systematic Review Centre for Laboratory Animal Experimentation’s (SYRCLE) risk of bias tool was used, which assessed domains including sequence generation, baseline characteristics, random housing, blinding, random outcome assessment, incomplete outcome data, and selective outcome reporting. For clinical studies, the Cochrane risk of bias tool (RoB 2.0) was applied, which evaluated five specific domains: bias arising from the randomization process, bias due to deviation from intended interventions, bias due to missing outcome data, bias in measurement of the outcome, and bias in the selection of the reported results. Following RoB 2.0 guidance, an overall risk of bias judgment (low, some concerns, or high) was derived for each specific outcome and for clinical and biochemical data.
The reporting of this study conforms to STROBE guidelines. 30 Artificial intelligence (AI) and/or AI-assisted technologies were not used during the preparation of this paper. Power considerations indicated that sample sizes were comparable to that of prior studies that detected medium between-group differences in macroergic compounds between nonpenetrating post-TBI and control data.
Results
Clinical characteristics
The general clinical patient group (n = 63) was aged between 22 and 61 years and included men (n = 36; 57.14%) and women (n = 27; 42.86%). The diagnosis of TBI was consistent with the International Classification of Functioning, Disability, and Health (T90.5, T90.0, S06.2, S06.3, and S06.0). Eligible participants were assigned to two clinical groups: the first consisted of 35 participants (55.56%) with a history of mild TBI in the anamnesis (group 1; mean age ± SD, 38.4 ± 12.98 years; range, 22–61) and the second comprised 28 participants (44.4%) with moderate TBI in the anamnesis (group 2; mean age ± SD, 35.82 ± 7.53 years; range, 24–51).
CT or MRI scans were performed in 32 participants from group 1 (91.43%) and revealed single vascular ischemic lesions (5.71% of patients), left-sided diffuse vascular injury (8.57%), pituitary stalk injury (14.28%), zones of swollen focal brain tissue in the temporal lobes (22.86%), and hydrocephalus (22.86%). CT and MRI scans were performed in all patients from group 2 and revealed dextral vascular lesions (3.57%), sinistral vascular lesions (7.14%), hydrocephalus (21.43%), focal lesions in cranial nerves and the pituitary stalk (28.57%), zones of swollen focal brain tissue in the frontal lobes (32.14%) or temporal lobes (7.14%), and a combination of findings (32.14%). Several participants in group 2 (57.14%) had mild neurological deficits, and 5 participants in group 2 had primary generalized tonic–clonic seizures (17.9%) with a seizure frequency of 1–2 per year and were not receiving antiepileptic drugs.
Study design
In this study, the functional state of oxidative phosphorylation and energy processes was assessed by measuring the levels of nucleotide adenines (ADP and ATP) in blood. The concentration of lactate, the final glycolysis product, was also determined as an indicator of hypoxia. 22 The concentration of pyruvate, the glycolysis intermediate product, was also measured; under anaerobic conditions it is converted to lactate via lactate dehydrogenase activation, and during aerobic conditions it is converted to acetyl coenzyme A (acetyl-CoA) by oxidative decarboxylation.15,22,29,31 Samples were collected at 1-year post-injury follow-up (12–14 months), representing the recovery period following nonpenetrating TBI. Patients were enrolled consecutively.
Biochemical data
This study found elevated CK levels in the general clinical group compared with HCs (p < 0.0001, Welch-corrected t = 7.030, 95% CI: −39.78 to −22.19, R = 0.4245). In the general clinical group, the median CK (mean ± SD) concentrations were 129.6 ± 21.37 IU/L at the 1-year post-injury follow-up (12–14 months) and 98.61 ± 19.74 IU/L in HCs (Table 1). Among these patients, we found that 14 (26.41%) showed an increase in CK levels of up to 34% of the median CK value. These findings should be interpreted with caution, as the physiology of increased CK is complex and multifactorial, 32 and the included patients represent a specific population; moreover, hidden somatic pathology may be present. In this study, mitochondrial oxidative disbalance together with low oxidative function was identified as a possible source of higher CK levels. We also showed improved outcome prediction in the post–mild TBI using CK levels. 22
Changes in the oxidative status in patients in the post-injury period following nonpenetrating traumatic brain injuries and healthy controls, M ± m.
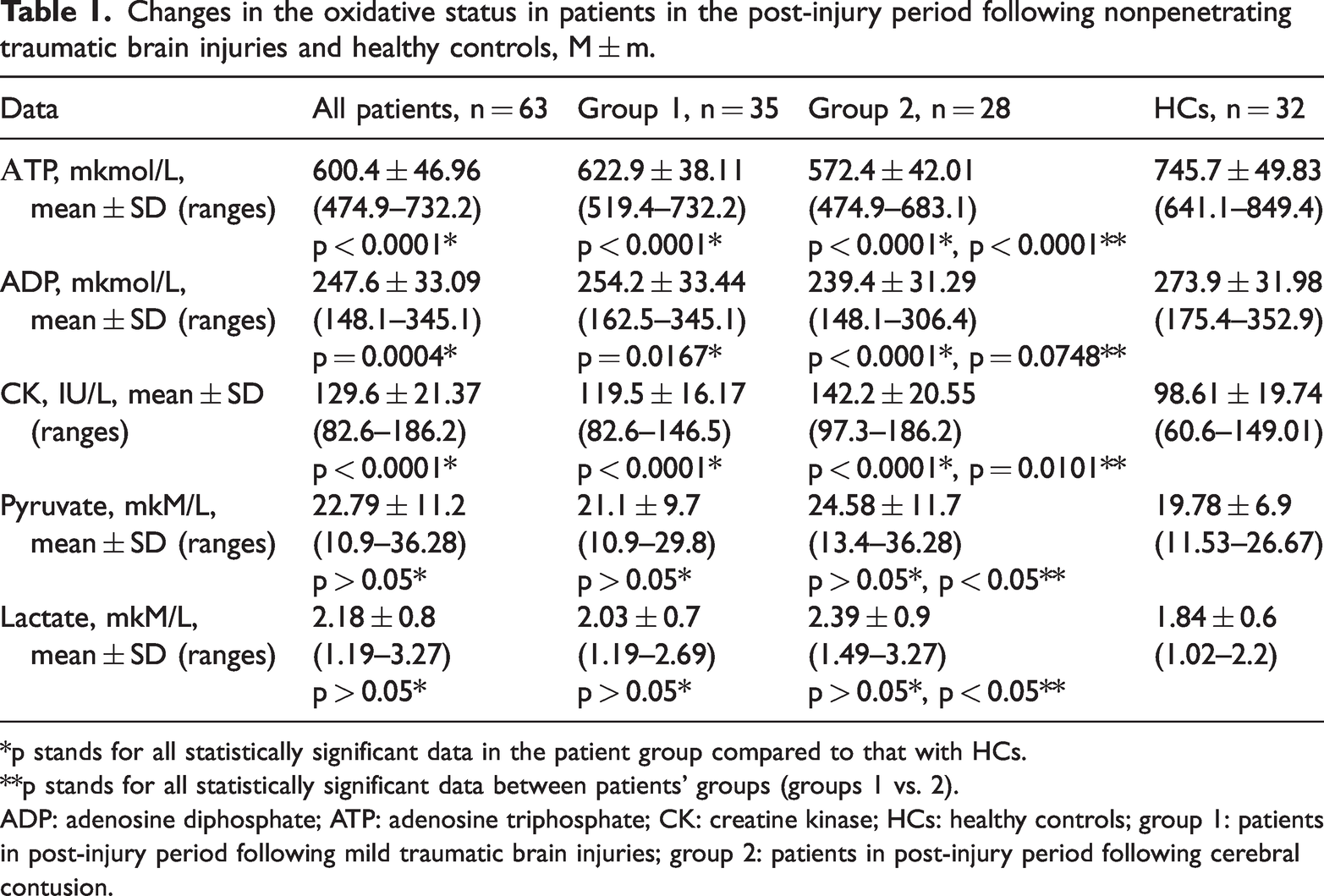
*p stands for all statistically significant data in the patient group compared to that with HCs.
**p stands for all statistically significant data between patients’ groups (groups 1 vs. 2).
ADP: adenosine diphosphate; ATP: adenosine triphosphate; CK: creatine kinase; HCs: healthy controls; group 1: patients in post-injury period following mild traumatic brain injuries; group 2: patients in post-injury period following cerebral contusion.
In this study, we also observed lower ATP levels in samples from the investigated participants than in HCs. In the general clinical group, the mean total ATP level was 600.4 ± 46.96 mkmol/L at 1-year post-injury follow-up, whereas it was 745.7 ± 49.83 mkmol/L in HCs (p < 0.0001, Welch-corrected t = 13.69, 95% CI: 124.0 to 166.5, R = 0.7597). Similarly, a decrease in ADP levels was also observed in these samples compared with HCs (p = 0.0004, Welch-corrected t = 3.734, 95% CI: 12.20 to 40.26); the median total ADP levels were 247.6 ± 33.09 mkmol/L in the general clinical group and 273.9 ± 31.98 mkmol/L in HCs (Table 1; Figures 1 and 2).

Adenosine triphosphate (mkmol/L) serum levels among patients in the post-injury period following nonpenetrating TBIs at 12–14 months post-injury follow-ups (general clinical group), following mild TBIs (group 1), and following cerebral contusion (group 2) as well as among HC. Error bars are mean ± SEM. *For all statistically significant data in the patients group compared with those for HC. **For all statistically significant data between patients group (groups 1 vs. 2). TBIs: traumatic brain injury; HC: healthy controls.

Adenosine diphosphate serum levels (mkmol/L) among patients in the post-injury period following nonpenetrating TBIs at 12–14 months post-injury follow-ups (general clinical group), following mild TBIs (group 1), and following cerebral contusion (group 2) as well as among HC. Error bars are mean ± SEM. *For all statistically significant data in the patients group compared with those of HC. **For all statistically significant data between patients group (groups 1 vs. 2). HC: healthy control; TBIs: traumatic brain injury.
To assess the prognostic and clinical relevance of ATP, ADP (Figures 1 and 2), and CK (Figure 3), data were divided according to the different forms of TBI. The mean CK and ATP levels were 119.5 ± 16.17 IU/L and 622.9 ± 38.11 mkmol/L, respectively, in patients from group 1, whereas they were 142.2 ± 20.55 IU/L and 572.4 ± 42.01 mkmol/L in patients from group 2. Higher CK (p < 0.0001, t = 4.779, 95% CI: 13.16 to 32.23, R = 0.03113) and lower ATP levels (p < 0.0001, t = 4.942, 95% CI: −71.01 to −30.04) were observed in patients after cerebral contusion (group 2) than those in patients from group 1 (Table 1; Figures 1 and 3). The mean ADP levels were 254.2 ±33.4 mkmol/L in patients from group 1, whereas it was 239.4 ± 31.29 mkmol/L in patients from group 2. Although the ADP levels were lower, the difference was not statistically significant (p = 0.0768, t = 1.814, 95% CI: −31.20 to 1.529).

CK (IU/L) serum levels among patients in the post-injury period following nonpenetrating TBIs at 12–14 months post-injury follow-ups (general clinical group), following mild TBIs (group 1), and following cerebral contusion (group 2) as well as among HC. Error bars are mean ± SEM. *For all statistically significant data in the patients group compared with those of HC. **For all statistically significant data between patients group (groups 1 vs. 2). HC: healthy control; TBIs: traumatic brain injury.
The present study demonstrated that CK, ATP, and ADP levels may serve as important metabolic biomarkers for monitoring oxidative stress–related deviances and oxidative dyshomeostasis during post-TBI follow-up. Brain meets its high energy requirements for rapid processing through metabolic processes of neurons and astrocytes.12,19,23,33
All observed biochemical changes in ATP–ADP and CK data were compared between patients from group 2 and HCs. The present study demonstrated that the concentrations of CK (p < 0.0001, t = 8.349, 95% CI −54.05 to −33.13, R2 = 0.5534), ATP (p < 0.0001, t = 14.62, 95% CI: 149.6 to 197.0, R2 = 0.7867), and ADP (p < 0.0001, t = 4.213, R2 = 0.2367, 95% CI: 18.09 to 50.85) were more pronounced in patients in the post-injury period following (S06.0; S06.2; S06.3) moderate TBI (Figure 4).
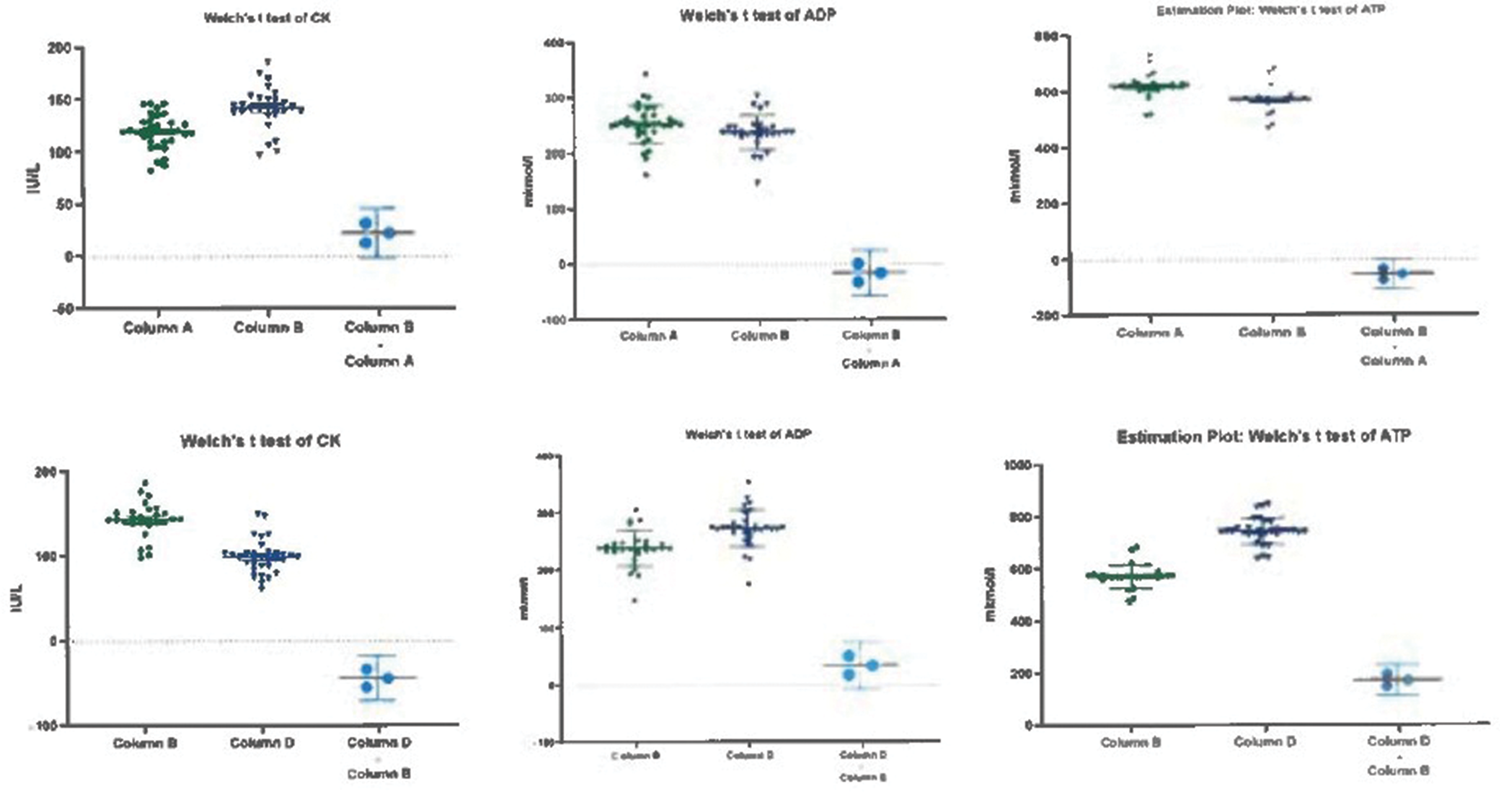
CK, ADP, and ATP data and unequal variances t-test in patients with mild post-TBIs (group 1, column A) versus post-TBIs following cerebral contusion (group 2, column B) and in post-TBIs following cerebral contusion (group 2, column B) versus HC (column D). ADP: adenosine diphosphate; ATP: adenosine triphosphate; CK: creatine kinase; HC: healthy control; post-TBIs: the post-injury period following TBIs.
As participants were not sex matched, statistically significant changes were compared between women and men. The concentrations of ATP, ADP, and CK were significantly different between female and male participants in the general clinical group (p = 0.188 for CK, p = 0.2 for ADP, and p = 0.262 for ATP) and among HCs. Additionally, no statistically significant differences were observed in lactate and pyruvate levels across all participants (p > 0.05). Although these levels were slightly elevated in patients with post-TBIs compared with HCs, the difference was not statistically significant; the median lactate level was 2.18 ± 0.8 mkM/L in patients and 1.84 ± 0.6 mkM/L in HCs. A similar pattern was observed for pyruvate, with median pyruvate level of 22.79 ± 11.2 mkM/L in patients and 19.78 ± 6.9 mkM/L in HCs. No differences in pyruvate and lactate levels were observed in group 2 compared with HCs (p > 0.05). These findings revealed no significant relationships between decreased ADP and ATP levels and lactate metabolic homeostasis. Future studies are required to clarify the role of lactate and ATP–ADP dyshomeostasis in the pathophysiology of nonpenetrating post-TBIs. Understanding metabolic homeostasis modulation will provide novel insights into potential therapeutic targets.
Discussion
The results of this study revealed the presence of definite oxidative metabolic dyshomeostasis in patients in the post-injury 1-year follow-up of nonpenetrating TBIs, and ATP (p < 0.0001, t = 13.69, 95% CI: 124.0 to 166.5) and ADP levels (p = 0.0004, t = 3.734, 95% CI: 12.20 to 40.26) remained low throughout the study period in all patient groups and were more pronounced in the post-injury period following contusion (S06.0; S06.2; S06.3). Herein, we also found elevated CK levels in the general clinical group compared with HCs (p < 0.0001, t = 7.030, 95% CI: −39.78 to −22.19), and these biochemical changes were more pronounced in the patients in the post-injury period following moderate TBI (S06.0; S06.2; S06.3). These findings suggest that in nonpenetrating post-TBIs, there was a severe deficiency of macroergic compounds, manifested by reduced ATP and ADP levels. Consequently, oxidative metabolic dyshomeostasis in these patients remains underrecognized and undertreated under baseline conditions. Despite their frequency and impact, there are no validated laboratory biomarkers to aid diagnosis, risk stratification, or treatment selection in nonpenetrating post-TBIs under baseline conditions during 12–14 months of post-injury follow-ups.
In this study, the pathogenetic role of low ATP and ADP in oxidative stress reactions contributing to the development of oxidative dyshomeostasis in nonpenetrating post-TBIs was emphasized under baseline conditions at 12–14 months post-injury follow-ups, representing the recovery period. This patient cohort may also show an increase in anaerobic glycolysis processes. We hypothesized that abnormal CK and lower ATP levels may serve as biomarkers for metabolic abnormalities in post-TBIs, particularly in moderate post-TBIs (S06.2; S06.3). Numerous clinical and experimental studies have demonstrated that post-TBIs result in oxidative stress and calcium dysregulation in mitochondria.13,16–18,23,27,28,31 Oxidative stress contributes to mitochondrial dysfunction during post-TBIs, and peripheral oxidative responses contribute to neuronal dysfunction after TBIs.17,27
In this study, the observed abnormal increase in CK and decrease in ATP and ADP serum levels across all patient groups may be associated with a decrease in the rate of glucose utilization by nervous tissue cells,4,5,16,17,20,21,24,26,27,34 which may be linked to underlying diseases. However, their role in post-TBIs remains controversial.
We observed higher CK levels (p < 0.0001, t = 4.779, 95% CI: 13.16 to 32.23) and lower ATP levels (p < 0.0001, t = 4.942, 95% CI: −71.01 to −30.04) after cerebral contusion than after mild TBIs at the 1-year post-injury follow-ups. Brain metabolism is maintained by neuronal and glial metabolic coupling, in which glial cells uptake glutamate from the synaptic cleft for future conversion and lactate production.5,6,11,22
We suggest that oxidative stress dysmetabolism plays an important role in the pathophysiology of the post-injury period following nonpenetrating moderate TBIs (S06.2; S06.3). Moreover, our results indicate that the post-injury period following cerebral contusion is associated with more inadequate and less effective enzyme metabolism.
Most studies describe changes in energy metabolism following TBIs as occurring in two phases: a period of hypermetabolism followed by prolonged hypometabolism in post-TBIs. 28 It is still not well understood how different cerebral metabolic states impact substrate metabolism and, ultimately, mitochondrial function. Based on the currently available data, this issue remains unresolved. Reperfusion exacerbates oxidative stress through a burst of reactive oxygen species.13,16,21,25,27,34,35 Moreover, evidence suggests that the less effective energy oxidative dysmetabolism following TBIs is associated with impaired glycolytic flux and oxidative phosphorylation, which are linked to disturbances in glucose transportation or its decreased utilization.
The nonpenetrating post-TBIs can lead to progressive neuronal dysfunction, with abnormal CK, ATP, and ADP levels partly reflecting complicated oxidative pathology. In contrast, the association with pyruvate and lactate levels showed no significant differences across groups.
The findings showed certain oxidative dyshomeostasis in nonpenetrating post-TBIs under baseline conditions at 12–14 months post-injury follow-ups. The oxidative stress imbalance was associated with mild abnormal changes in CK, ADP, and ATP levels. These variations were more related to the post-injury period after cerebral contusion compared with HC (for ADP: p < 0.0001, t = 4.213, 95% CI: 18.09 to 50.85; for ATP: p < 0.0001, t = 14.62; 95% CI: 149.6 to 197.0; for CK: p < 0.0001, t = 8.349; 95% CI: −54.05 to −33.13), with higher CK (p < 0.0001, t = 4.779, 95% CI: 13.16 to 32.23) and lower ATP levels (p < 0.0001, t = 4.942, 95% CI: −71.01 to −30.04) in the patients after cerebral contusion than those in group 1. The study also showed the critical need for continued investigations of other related biomarkers of oxidative status and related enzymes involved in energy-dependent biochemical processes in patients with post-TBIs, for deeper evaluation of different pathogenetic mechanisms and future treatment options for this patient cohort.10,33
The findings demonstrated a certain oxidative stress imbalance in the post-injury period, with oxidative stress associated with higher CK and lower ATP serum levels, and these changes were more related to moderate TBIs.
The abnormalities associated with identified oxidative metabolic dyshomeostasis may provide different potential antioxidant therapeutic strategies for normalizing and controlling oxidative metabolism in patients with post-TBIs. This investigation provides a framework in which observations regarding systemic biomarkers involved in oxidative metabolism and oxidative stress reaction generation can be interpreted, and it aligns with diverse fields of priority for subsequent and future investigations in patients with post-TBIs.
This study has several strengths. In the post-injury period following nonpenetrating TBIs, definite oxidative metabolic dyshomeostasis was observed, with higher CK levels associated with lower ATP–ADP levels at 12 months post-injury follow-ups. These changes were more pronounced in patients who experienced after brain contusion.
This study provides important insights into the pathogenetic role of systemic enzymes involved in oxidative metabolism and oxidative stress reaction generation, emphasizing their contribution to the development of oxidative dyshomeostasis in nonpenetrating post-TBIs and covering future pathogenetic treatment options.
Advancements in the diagnostics of oxidative metabolic dyshomeostasis in nonpenetrating post-TBIs are critical for any future research, clinical outcomes, and progression of post-TBIs. Despite the prevalence and impact of metabolic dyshomeostasis, there are no validated laboratory biomarkers to aid diagnosis, risk stratification, or treatment selection in these patients. The development of new biomarkers of metabolic dyshomeostasis can provide deeper insights into the pathogenesis of post-TBIs and the effectiveness of treatments, thereby guiding effective therapeutic interventions. Having deeper insights into the long-term effects of post-TBIs remains crucial for future research. Longitudinal studies are needed to investigate how oxidative metabolic dyshomeostasis in nonpenetrating post-TBIs evolves over time and influences post-TBIs progression. Future pharmacological research should focus on integrative treatment strategies that modulate oxidative metabolic dyshomeostasis, which would not only target post-TBIs symptoms but also address long-term recovery and improve patient outcomes.
Limitations of the study
The following limitations should be considered when interpreting these results. During the investigation, blood and cerebrospinal fluid samples from participants with acute TBI along with neuroimaging data, including details of blood–brain barrier breakage during TBI structure were unavailable. Obtaining these data is particularly challenging in this special patient cohort during the acute TBI phase. Enzymes involved in cellular energy metabolism are known to vary with blood sample timing and handling, which may have restricted the analysis.
This study excluded patients who were regularly taking psychiatric and antiepileptic drugs to rule out the potential effect of these medications on the biochemistry of enzyme levels. Thus, patients with severe post-TBI forms were not included in this analysis. Longer-term studies are needed to assess outcomes in patients with nonpenetrating TBIs and for those not receiving treatment. The number of these patients should be increased, possibly through multicentric studies. Although the results are promising, additional clinical validation and in vivo experiments are needed to confirm relationships between metabolic-related parameters, EDSS, and clinical differences. Additionally, the dataset represents only the Ukrainian population. Moreover, these limitations did not lower any scientific values in the investigation.
Conclusions
The nonpenetrating post-TBIs at 12–14 months post-injury follow-ups, representing the time of recovery, showed abnormally higher median total CK levels associated with lower total ATP and ADP levels. This indicates a definite baseline oxidative metabolic dyshomeostasis, which may play an important role in pathogenetic mechanisms contributing to the development of long-term consequences of TBIs and was more pronounced in patients after cerebral contusions.
Footnotes
Acknowledgments
The author is very grateful to Gorbatch T.V., Ph.D. (Department of Biochemistry, Kharkiv National Medical University), for amazing biochemical support during the investigation. The author is very grateful to Gubina-Vakulik Galina, PhD, M.D., Professor (Department of Pathomorphology, Kharkiv National Medical University), for the provision of study materials, reagents, and other biochemical materials. The author thanks enormously Posokhov M.F., PhD, M.D. (the Head of the Department of Neurosurgery and Paroxysmal States, INPN NAMSU) for data analysis assistance and some patients’ follow-up.
Author contributions statement
The study conception, design, data collection, data analysis, and interpretation of the obtained results were performed by Yevgeniya Lekomtseva. Andrey Belousov contributed to the study by providing additional statistics and by reviewing and editing the revised publication. Vitaliy Tkachenko was responsible for writing, calculating the additional statistical results and methodology, and reviewing during revision. All authors have reviewed and edited the content as needed, took full responsibility for the content of the publication, and approved the final manuscript. The authors did not use the AI service during the writing of this paper.
Data availability statement
Anonymized data will be available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declare no conflicts to disclose; the authors have no financial conflicts of interest. All authors confirm that they have read the Journal’s position on issues involved in ethical publications and affirm that this research is consistent with its guidelines.
Ethical approval
This study was conducted in accordance with the Declaration of Helsinki and was approved by the local health ethics committee of the State Institute of Neurology, Psychiatry, and Narcology of the Academy of National Medical Sciences of Ukraine, Kharkiv, Ukraine (protocol code: 70-O and date of approval: 04 February 2016).
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or nonprofit sectors.