
Abstract
Objective
This study aimed to investigate the protective effects of β-escin against neuroinflammatory injury and its influence on the interleukin-6/Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway in a rat model of ischemic stroke.
Methods
Rats underwent a 2-h middle cerebral artery occlusion and were categorized into sham, middle cerebral artery occlusion, and middle cerebral artery occlusion treated with β-escin (0.45, 0.90, and 1.80 mg/kg) groups. Cerebral damage was assessed using 2,3,5-triphenyltetrazolium chloride and hematoxylin and eosin staining; neuronal apoptosis was evaluated via terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay; serum levels of inflammatory cytokines (interleukin-6, tumor necrosis factor-α, and interleukin-1β) were measured using enzyme-linked immunosorbent assay, and protein expression of the interleukin-6/Janus kinase 2/signal transducer and activator of transcription 3 pathway was analyzed using western blot.
Results
β-escin administration dose-dependently reduced Longa scores, cerebral infarction volume, and pathological damage while also attenuating neuronal apoptosis. It significantly suppressed the release of proinflammatory cytokines and downregulated the expression of interleuin-6 and interleukin-6 receptor as well as the ratios of phosphorylated Janus Kinase 2/Janus Kinase 2 and phosphorylated signal transducer and activator of transcription 3/ signal transducer and activator of transcription 3. These protective effects were positively correlated with the dosage of β-escin.
Conclusion
The findings suggested that β-escin exerted neuroprotective effects in ischemic stroke by modulating the interleukin-6/Janus kinase 2/signal transducer and activator of transcription 3 pathway, thereby reducing neuroinflammation and apoptosis.
Keywords
Introduction
Stroke is the second leading cause of death worldwide, following cardiovascular diseases; however, treatment options for this condition remain limited. Among stroke subtypes, ischemic strokes account for approximately 87% of cases, predominantly resulting from cerebral vascular occlusion or rupture due to impaired blood flow. 1 Recent studies have suggested that the immunoinflammatory response triggered by ischemia–reperfusion injury plays a critical role in determining disease progression and clinical outcomes. 2 Despite decades of research focused on revascularization and neuronal protection as primary treatment approaches, 3 only a few pharmacological agents have been successfully developed, all of which are constrained by a narrow therapeutic window (4–6 h post-onset). This limitation highlights the urgent need for novel therapeutic targets and intervention approaches. Notably, cerebral ischemia induces neuronal apoptosis and necrosis, initiating a cascade of inflammatory responses and intricate pathophysiological alterations.
Numerous studies have identified inflammation as a central feature of brain injury during ischemic stroke and reperfusion.4–6 The interleukin (IL)-6/Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) signaling pathway is a key inflammatory cascade involved in immune regulation, cytokine production, and adaptive response activation, contributing significantly to cytokine-mediated inflammation.7,8 This pathway also plays an important role in mediating tissue cell injury after cerebral ischemia by regulating inflammatory cytokine responses and influencing apoptosis in ischemic brain cells. 9 Tian et al. 10 demonstrated that inhibition of the JAK2/STAT3 signaling pathway can reduce cerebral infarction lesions and improve neurological function following middle cerebral artery occlusion (MCAO).
Escin, the primary active component derived from Aesculus chinensis Bunge, is a pentacyclic triterpenoid saponin existing in two isomeric forms, α and β, which demonstrates distinct physical and chemical properties. Among these, β-escin, the more biologically active and widely utilized isomer, exhibits a range of pharmacological effects, including antiexudation, anti-inflammatory, antiswelling, and antitumor activities. It also promotes blood circulation, improves venous tone, and facilitates the clearance of reactive oxygen species.11–14 Clinically, escin is used in the management of chronic venous insufficiency, various forms of edemas, and certain tumors. Additionally, β-escin has demonstrated neuroprotective effects against cerebral ischemia–reperfusion injury, reducing local inflammation and infarct size.15–17 Currently, no studies have reported the specific mechanism through which β-escin affects the inflammatory response in ischemic stroke. This study investigated the impact of β-escin on the IL-6/JAK2/STAT3 signaling pathway in a rat model of ischemic stroke, aiming to elucidate the mechanism by which β-escin ameliorates ischemic stroke. The findings offer a valuable experimental foundation for the further development and clinical application of this drug.
Materials and methods
Animal & study design
The experiment employed 100 specific pathogen-free (SPF) grade male Sprague–Dawley rats, weighing between 220 and 250 g, which were procured from Hunan Slack Jingda Laboratory Animal Co., Ltd. The animals were kept under controlled environmental conditions at 25°C ± 2°C with a 12-hour light–dark cycle and were provided with ad libitum access to standard rodent feed and water. This study involved only animal models. The experimental protocol was reviewed and officially approved by the Animal Ethics Committee of The Fourth Hospital of Changsha, affiliated with Hunan Normal University, which confirmed that the requirements for human subject research ethics approval were not applicable. All procedures were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and formally approved under the ethical approval number 2023-062.
Prior to the surgical procedure, all rats were subjected to a 12-h fasting period, with water withheld. Subsequently, they were randomly divided into five groups according to body weight: sham operation group (sham, n = 10), MCAO group (n = 22), and three MCAO groups treated with β-escin (≥95%, powder; Sigma-Aldrich, Cat# A5889; USA) at doses of 0.45 mg/kg (n = 24), 0.90 mg/kg (n = 22), and 1.80 mg/kg (n = 22). These doses of β-escin were selected based on our preliminary experiments and with reference to recent studies employing similar parenteral administration routes in ischemic stroke models.16,18 Rats in the treatment groups received intraperitoneal injections of the corresponding dose of β-escin immediately after modeling, and the administrations were continued once daily for 7 consecutive days. Meanwhile, rats in the MCAO and sham groups were injected intraperitoneally with an equivalent volume of normal saline at the same points (Figure 1). All rats were euthanized on the 8th day following surgery using an overdose of ketamine and xylazine in a 7:3 ratio.

Schematic timeline of the events of the study.
MCAO model
The middle cerebral artery (MCA) is commonly implicated in cerebral ischemic injuries; therefore, the MCAO model is widely employed in studies of focal cerebral ischemia. 19 In this study, ischemic stroke was induced in rats using the MCAO method, following a previously established protocol. Briefly, rats were anesthetized with a 1% pentobarbital solution (50 mg/kg), and the right common carotid artery was surgically exposed. Subsequently, a nylon monofilament was introduced to occlude the middle cerebral artery via the right common carotid artery. After 2 h of ischemia, the filament was withdrawn to allow reperfusion, which occurred through collateral circulation originating from the contralateral internal carotid artery, vertebrobasilar artery, and cortical branches of the cerebral artery via the basilar arterial ring. In the sham group, the same surgical procedure was performed without insertion of the nylon filament.
Assessment of neurological impairment
Neurological impairment was evaluated using the Longa scoring system. 20 The scoring criteria were as follows: 0, no observable neurological deficits; 1, contralateral forelimb flexion without full extension; 2, mild circling toward the opposite side; 3, severe circling or falling to the opposite side; and 4, contralateral paralysis and inability to walk spontaneously. Higher scores corresponded to more severe neurological impairments.
2,3,5-triphenyltetrazolium chloride (TTC) staining
After neurological evaluation, TTC staining was performed to quantify the cerebral infarction volume. 21 Selected rats from each group were anesthetized, and blood samples were collected via the abdominal aorta. Brain tissue was rapidly harvested, frozen at −20°C for 30 min, and sectioned coronally into six consecutive slices of 2-mm thickness. The slices were immersed in TTC staining solution (which was prepared in-house as follows: 2 g of TTC (Sigma-Aldrich) was dissolved in 100 mL of phosphate-buffered saline (PBS), after which the solution was filtered for sterilization and stored away from light) and incubated at 37°C in the dark for 30 min. After staining, all sections were fixed in 4% paraformaldehyde for 24 h and subsequently photographed for further analysis. The infarct area was assessed using Image-Pro Plus software 6.0 (Media Cybernetics Inc., USA), and the cerebral infarct volume ratio was calculated as the percentage of the total pale infarct area relative to the total area of the brain sections.
Hematoxylin and eosin (H&E) staining
Rats were perfused transcardially with 4% paraformaldehyde solution. The brains were then carefully removed and immersed in 4% paraformaldehyde for 48 h for fixation. Subsequently, the tissues were embedded in paraffin and sectioned at a thickness of 6 μm. The sections were stained with H&E according to standard protocols. 6 Pathological alterations in the ischemic penumbra region of the brain were examined and imaged under an optical microscope (OLYMPUS IX71, Japan).
TdT-mediated dUTP nick end labeling (TUNEL) staining
Apoptosis was evaluated using a TUNEL assay kit (Beyotime Biotechnology, Beijing, China) on paraffin-embedded brain sections. 22 After deparaffinization and rehydration, the sections were treated with 20 μg/mL proteinase K solution for 30 min at 37°C to achieve permeabilization. The sections were then incubated in a humidified chamber with terminal deoxynucleotidyl transferase (TdT) and a fluorescein-labeled 2′-deoxyuridine, 5′-triphosphate (dUTP) solution for 1 h at 37°C. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Images were captured using a fluorescence microscope, and the number of TUNEL-positive neurons was quantified using Image-Pro Plus software version 6.0.
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were centrifuged at 3000 r/min for 15 min to separate the serum. Levels of IL-6, tumor necrosis factor-α (TNF-α), and IL-1β were measured using commercially available ELISA kits (Rat Tumor Necrosis Factor ELISA Kit, Cat. No. RAB0479; Rat IL-6 ELISA Kit, Cat. No.RAB0311; R&D Systems, USA) according to the manufacturer’s instructions. Absorbance was recorded at 450 nm, and cytokine concentrations were determined based on a standard curve.
Western blotting assay
Protein expression levels of IL-6, IL-6 receptor (IL-6R), JAK2, phosphorylated-JAK2 (p-JAK2), STAT3, and phosphorylated STAT3 (p-STAT3) were analyzed using western blot. Brain tissues were homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer containing phenylmethylsulfonyl fluoride and phosphatase inhibitors. Total protein concentration was quantified using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, USA). Equal amounts of protein were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes. The membranes were blocked and incubated overnight at 4°C with the following primary antibodies: anti-IL-6, anti-IL-6R, anti-JAK2, anti-p-JAK2, anti-STAT3, anti-p-STAT3 (all from Aifang Biotech), and anti-β-actin (Immunoway). After washing with Tris buffered saline with Tween 20 (TBST), the membranes were incubated with horseradish peroxidase (HRP)–conjugated secondary antibodies. Protein bands were visualized using an enhanced chemiluminescence (ECL) detection system and exposed to X-ray film. Band densities were analyzed using BandScan software (BandScan 4.0, Glyko), with β-actin serving as the loading control. Each experiment was repeated four times (n = 4) using independent biological replicates.
Data analysis and statistics
All statistical analyses were performed using GraphPad Prism 9.5 (GraphPad Software, USA) and IBM Statistical Package for Social Sciences (SPSS; version 26.0, IBM Corp., USA). Quantitative data were expressed as mean ± SD. Comparisons among multiple groups were conducted using one-way analysis of variance (ANOVA), followed by appropriate post hoc tests where applicable. A p-value of ≤0.05 was considered statistically significant.
Results
β-escin provides neurological deficit protection in MCAO rats
As illustrated in Figure 2, β-escin significantly improved neurological function in a rat model of ischemic stroke. Neurological assessment was performed using the Longa five-point scoring system. The sham group exhibited a lower neurological impairment score, whereas the MCAO group demonstrated a significantly higher Longa score of 3.57 ± 0.53 (P < 0.01) than the β-escin (1.80 mg/kg) dose group (P < 0.01). With the decrease in the dose of β-escin administration in rats, the score gradually increased.

β-escin provides neurological deficit protection in MCAO rats. Data are presented as mean ± SD. **P < 0.01 compared with the sham group; ##P < 0.01 compared with the MCAO group; n = 10 for each group. MCAO: middle cerebral artery occlusion.
β-escin effectively decreases cerebral infarction volume in MCAO rats
As shown in Figure 3, β-escin treatment significantly attenuated the cerebral infarction volume in MCAO rats. TTC staining revealed uniformly red-colored brain tissue with no detectable infarction in sham-operated rats. In contrast, MCAO control rats exhibited distinct white infarct areas occupying 36.98% ± 2.71% of total brain volume (P < 0.01 vs. sham). Administration of 1.80 mg/kg β-escin significantly reduced the infarct area to 29.05% ± 2.92% (P < 0.05 vs. MCAO), providing further evidence for its neuroprotective properties.

Illustration of the impact of β-escin on reducing the cerebral infarction volume in rats subjected to MCAO. (a) Representative TTC-stained coronal brain sections and (b) quantitative analysis of infarct volume expressed as percentage of the total brain volume. Data are presented as mean ± SD. **P < 0.01 compared with the sham group; #P < 0.05 compared with the MCAO group. MCAO: middle cerebral artery occlusion; TTC: 2,3,5-triphenyltetrazolium chloride.
β-escin can improve the pathological changes of the brain penumbra region of MCAO rats
As demonstrated in Figure 4, H&E staining revealed neatly arranged brain neuronal cells with regular shapes and clear, complete nuclei in the sham group. In contrast, in the MCAO group, the core area of cerebral ischemia exhibited a significant reduction in the neuronal cell count (P < 0.01), with scattered arrangement, unclear structure, condensed and variably sized nuclei, and a few cells undergoing degeneration and necrosis. Notably, β-escin demonstrated a dose-dependent enhancement in brain tissue pathology in rats compared with that in the MCAO group, with a significant increase in the neuronal count in the medium- and high-dose groups (P < 0.01), and partial recovery of nuclear structures in the cortical ischemic penumbra. These results suggested that β-escin exerted neuroprotective effects against MCAO-induced cerebral damage.

H&E staining of rat brain. (a) Morphological structure of cells under optical microscope (200× and 400×; scale 200 and 100 μm) and (b) survival number of neurons. **P < 0.01 compared with the sham group; ##P < 0.01 compared with the MCAO group. H&E: hematoxylin and eosin; MCAO: middle cerebral artery occlusion.
β-escin mitigates neuronal apoptosis in the ischemic penumbra of MCAO rats
As demonstrated in Figure 5, TUNEL staining revealed significantly enhanced neuronal apoptosis in the ischemic region. Compared with the sham group, the MCAO group exhibited a significant increase in TUNEL-positive neurons and cell numbers in rat brain tissue (P < 0.01). β-escin administration dose-dependently reduced the apoptotic response as follows: medium-dose treatment decreased TUNEL-positive neuron counts versus MCAO controls (P < 0.05) and high-dose treatment demonstrated more pronounced reduction (P < 0.01). These findings suggested that β-escin exerted antiapoptotic effects in cerebral ischemia.

Inhibitory effect of β-escin on neuronal apoptosis in MCAO rats. (a) TUNEL staining showed that apoptotic cells were red, and DAPI staining showed that the nucleus was blue (200×; scale 200 μm) and (b) apoptosis percentage of TUNEL-positive neurons. Statistical significance is indicated as follows: **P < 0.01 compared with the sham group and #P < 0.05 and ##P < 0.01 compared with the MCAO group. MCAO: middle cerebral artery occlusion; TUNEL: TdT-mediated dUTP nick end labeling; DAPI: 4′,6-diamidino-2-phenylindol.
β-escin suppresses proinflammatory cytokine release in MCAO rats
We employed ELISA method to measure serum levels of proinflammatory factors, including IL-6, TNF-α, and IL-1β in rats, which are indicative of inflammatory responses induced by cerebral ischemia and reperfusion. Figure 6 illustrates that serum levels of IL-6, TNF-α, and IL-1β in the MCAO group were significantly elevated at 100.68 ± 4.22 pg/mL, 266.34 ± 19.27 pg/mL, and 30.68 ± 4.48 pg/mL (P < 0.01), respectively, compared with those in the sham group. In the β-escin middle- and high-dose groups, serum levels of IL-6, TNF-α, and IL-1β were significantly reduced (P < 0.05 and P < 0.01). These results suggested that β-escin intervention in MCAO exerted a neuroprotective effect by inhibiting the inflammatory response.
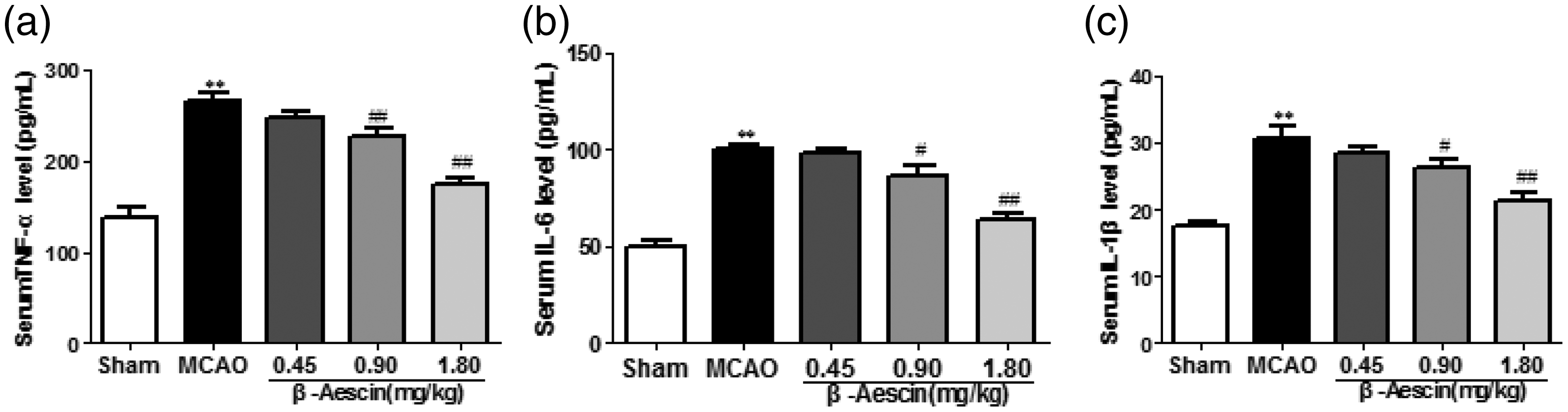
Illustration of the impact of β-escin on proinflammatory cytokine levels, particularly IL-6, TNF-α, and IL-1β in MCAO rats. *P < 0.05 and **P < 0.01 compared with the sham group; #P < 0.05 and ##P < 0.01 compared with the MCAO group. MCAO: middle cerebral artery occlusion; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α; IL-1β: interleukin-1β.
β-escin influences the expression of proteins involved in the IL-6/JAK2/STAT3 signaling pathway
The IL-6/JAK2/STAT3 pathway plays a critical role in amplifying inflammatory cytokine release, thereby exacerbating neurological damage. To investigate the mechanism underlying neuroprotective effects of β-escin, we analyzed key pathway components (IL-6, IL-6R, JAK2, p-JAK2, STAT3, and p-STAT3) via western blotting. Figure 7 illustrates a significant increase in the relative expression levels of IL-6, IL-6R, p-JAK2/JAK2, and p-STAT3/STAT3 in the MCAO group compared with those in the sham group (P < 0.01). Following β-escin treatment at doses of 0.90 and 1.80 mg/kg, the relative expression levels of IL-6, IL-6R, p-JAK2/JAK2, and p-STAT3/STAT3 were significantly decreased, showing a positive dose-response relationship (P < 0.05 and P < 0.01). These results demonstrated that β-escin attenuated neuroinflammation by targeting the IL-6/JAK2/STAT3 cascade, suggesting a potential therapeutic mechanism for ischemic brain injury.

Illustration of the western blot analysis and relative expression levels associated with the β-escin-mediated IL-6/JAK2/STAT3 signaling pathway. (a) IL-6 and IL-6R protein bands and relative expressions; (b) p-JAK2/JAK2 protein bands and ratios; and (c) p-STAT3/STAT3 protein bands and ratios. **P < 0.01 compared with the sham group; #P < 0.05 and ##P < 0.01 compared with the MCAO group. MCAO: middle cerebral artery occlusion; IL-6: interleukin-6; JAK2: Janus kinase 2; STAT3: signal transducer and activator of transcription 3; IL-6R: IL-6 receptor; p-JAK2: phosphorylated Janus kinase 2; p-STAT3: phosphorylated signal transducer and activator of transcription 3.
Discussion
Although the mortality rates associated with ischemic stroke have gradually declined in recent years, the persistently high prevalence and resulting disability continue to impose substantial socioeconomic burdens worldwide. Following cerebral ischemia, neurons in the infarct core undergo rapid apoptotic and necrotic processes. Timely restoration of blood flow to the affected area can salvage the surrounding penumbral tissue, thereby minimizing neurological deficits. 23 The ischemic penumbra exhibits a distinct temporal pattern of cell death, with progressive deterioration occurring over hours to days post-stroke. This region represents a critical therapeutic target, as its potential reversible damage offers a window for intervention. 24 However, prolonged ischemia leads to hypoxia and energy depletion in the penumbra, ultimately resulting in irreversible injury. Recent advances combining endovascular thrombectomy with tissue plasminogen activator have significantly improved outcomes in patients with ischemic stroke. 25 Nevertheless, this treatment remains constrained by a narrow therapeutic window (≤4.5 h post-onset), as thrombolytic therapy beyond this period may cause reperfusion injury. Notably, <50% of patients regain functional independence and approximately 10% achieve full neurological recovery. 26 Therefore, the main goals of ischemic stroke treatment are to promote vascular recanalization, reduce apoptosis of nerve cells, and alleviate damage to the ischemic penumbra. 27 The development of a neuroprotective agent with a clear mechanism of action and significant efficacy to alleviate the damage of the ischemic penumbra has become the focus of the current research.
Mounting evidence establishes inflammation as a critical mediator in ischemic stroke pathology. This process involves the production of inflammatory mediators and immune cell chemotaxis, which collectively drive the inflammatory cascade. Subsequently, inflammatory cytokines facilitate leukocyte infiltration into neural tissue. This triggers microglial activation, amplifying production of proinflammatory factors, which in turn promotes neuroexcitotoxicity, oxidative stress, and apoptotic/necrotic pathways. These mechanisms ultimately induce cerebral edema, hemorrhage, and amplify neural damage through secondary neuronal death.28–30 Neuroprotective agents can mitigate ischemic brain injury by suppressing inflammation in nerve tissue, with targeting specific inflammatory cytokines offering notable therapeutic potential. 31
In ischemic stroke, excessive reactive oxygen species (ROS) activate the inflammatory cascade via the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway. NF-κB, typically existing in the form of p65–p50 dimers that binds to the inhibitory protein IκBα, is retained in the cytoplasm. 32 Following ROS stimulation, IκBα is phosphorylated, resulting in its translocation and enhanced nuclear translocation of NF-κB, which upregulates NF-κB p65 mRNA expression and activates microglia. These activated microglia release proinflammatory cytokines, including IL-1β, IL-6, IL-8, and TNF-α, which regulate immune responses, cell proliferation, and differentiation, thereby exacerbating cerebral ischemia-induced brain injury. Using the MCAO model to simulate ischemic stroke, we observed that neurological deficits and cerebral infarction volume (assessed via Longa score, H&E staining, and TTC staining) were exacerbated, accompanied by increased platelet aggregation, blood viscosity, and thrombosis. β-escin significantly attenuated cell damage in ischemic penumbra and provided neuroprotective effects. ELISA and TUNEL staining demonstrated that β-escin markedly reduced inflammatory cytokines (IL-1β, IL-6, and TNF-α) and neuronal apoptosis, findings consistent with clinical reports. Li et al. 33 reported that β-escin modulates NF-κB content in the intestinal barrier during cerebral ischemia, whereas Hu et al. 17 reported that β-escin significantly inhibits apoptosis following transient focal cerebral ischemia in rats. Our research is consistent with their report and, moreover, it further enriches the theoretical basis of the mechanism of β-escin in treating cerebral ischemia.
IL-6, a 212-amino acid phosphorylated glycoprotein, functions as a pleiotropic inflammatory mediator. During cerebral ischemia, IL-6 expression becomes markedly elevated, stimulating vascular endothelial cells to upregulate adhesion molecule production. This process facilitates extensive leukocyte recruitment and aggregation at the injury site, thereby amplifying the inflammatory cascade and exacerbating ischemic brain damage. 34 Therefore, IL-6 is an important indicator of the occurrence, development, prognosis, and signaling pathways of ischemic stroke. 35 JAK2, a Janus kinase family member prevalent in brain tissue, can be inhibited to reduce cyclin D1 expression and neuronal apoptosis. 36 STAT3, part of the STAT family, is prevalent in the central nervous system. It facilitates direct signal transmission to the nucleus, enhancing the transcription and expression of genes encoding proinflammatory mediators such as cytokines, chemokines, adhesion molecules, and inflammatory enzymes. The neuroinflammatory response after cerebral ischemia involves abnormal STAT3 activation.37,38 IL-6, which is transiently upregulated, interacts with target cells via IL-6R and forms a signaling complex with the coreceptor glycoprotein 130 (gp130) homodimer. This complex activates JAK-associated kinase signaling and STAT3, playing a crucial role in inflammation. Research has indicated that JAK2/STAT3 signaling is activated early in cerebral ischemia, contributing to oxidative stress, inflammatory responses, and neuronal apoptosis. Inhibition of p-JAK2 and p-STAT3 expression provides neuroprotective benefits.39,40 Luo et al. 41 demonstrated that bazedoxifene significantly reduces inflammation and offers therapeutic benefits for cardiovascular disease by inhibiting the IL-6/IL-6R/STAT3 signaling pathway. Western blot analysis revealed that β-escin significantly modulates the IL-6/JAK2/STAT3 signaling pathway proteins in the brains of rats with ischemic stroke, contributing to neuroprotection by regulating neuroinflammation.
Our study provided a new therapeutic approach for β-escin as a treatment for ischemic injury. This study has several limitations: First, the research was conducted exclusively on a rat model, whose pathological mechanisms differ from those of human diseases. Therefore, the clinical translatability of the study outcomes requires further validation through subsequent human tissue studies or clinical trials. Second, although this study focused on the specific IL6/JAK2/STAT3 signaling axis, it did not explore the potential involvement of other related pathways, such as NF-κB or mitogen-activated protein kinase signaling. Future research should expand the analysis to include interactions among multiple pathways. Finally, the pharmacokinetic properties of β-escin, such as blood–brain barrier penetration and in vivo metabolic processes, have not been fully evaluated and require further investigation through subsequent pharmacological studies.
Conclusion
This study explored the neuroprotective mechanism of β-escin in a rat model of ischemic stroke. The study demonstrated that β-escin effectively mitigated neurological deficits, decreased cerebral infarction volume, and ameliorated pathological damage in ischemic stroke. These outcomes were likely associated with the modulation of the IL-6/JAK2/STAT3 neuroinflammatory signaling pathway as well as the suppression of brain inflammation and apoptosis.
Footnotes
Acknowledgments
This research received funding from the Changsha Natural Science Foundation of China (Grant number, kq2202028).
Acknowledgment of the use of generative AI and AI-assisted technologies in the writing process
The authors did not utilize any generative AI tools, including ChatGPT, NovelAI, Jasper AI, Rytr AI, or DALL-E, during the preparation of this work.
Author contributions
Duan Yanqin: Conceptualization, Methodology, Investigation, Formal analysis, and Writing–Original Draft. Liu Shenglan: Validation, Investigation, and Data Curation. Zhang Yu: Resources and Software. Deng Yu: Supervision and Project administration. Du Qing: Visualization and Writing–Review & Editing. Zeng Hongliang: Validation and Investigation. Tan Dianbo: Resources. Liu Chunhai: Supervision. Yin Yong: Funding acquisition. Liu Dongliang: Conceptualization, Resources, Writing–Review & Editing, Supervision, and Funding acquisition.
Data availability statement
The study’s data can be obtained from the corresponding author upon reasonable request.
Declaration of competing interest
The authors state that no financial interests or personal relationships exist that could have influenced the work presented in this paper.
Funding
This research received funding from the Changsha Natural Science Foundation of China under grant number kq2202028.