
Abstract
Objective
Emerging evidence has indicated the potential role of DNA damage response in asthma pathogenesis, but the underlying mechanisms remain elusive. Therefore, this study aimed to identify key diagnostic DNA damage response–related genes in asthma and explore their regulatory networks.
Methods
Differentially expressed genes between healthy individuals and patients with asthma were identified using the Gene Expression Omnibus database. Hub DNA damage response–related differentially expressed genes were determined via protein–protein interaction network and verified through gene expression analysis. Receiver operating characteristic curve was employed to identify diagnostic genes. Transcription factor–microRNA–target gene interactions were analyzed to uncover the regulatory networks in asthma pathogenesis. In this observational study, reverse transcription quantitative polymerase chain reaction was used to validate gene expression levels in healthy individuals and patients with asthma.
Results
Six of the nine hub genes (ATM, PCNA, CUL4A, PARP2, HLTF, and NBN) were identified as key diagnostic genes. These genes may contribute to asthma progression by regulating inflammatory pathways, such as cyclic GMP–AMP synthase–stimulator of interferon genes, senescence-associated secretory phenotype, autophagy, and apoptosis. Three microRNAs and eleven transcription factors were recognized as potential regulators. Reverse transcription quantitative polymerase chain reaction confirmed the downregulation of DNA damage response genes in asthma and revealed distinct expression patterns across different asthma endotypes.
Conclusion
Six DNA damage response–related genes may serve as diagnostic biomarkers for asthma, and the transcription factor–microRNA–DNA damage response gene network highlights the role of DNA damage response in asthmatic inflammation.
Keywords
Introduction
Asthma is a chronic inflammatory respiratory disease characterized by airway hyperresponsiveness and remodeling. Although the age-standardized rates of asthma burden have decreased over the past three decades, the overall burden of asthma remains substantial and continues to rise. Globally, 262.41 million prevalent cases (95% uncertainty level: 224.05 to 309.45) have been reported. 1 Generally, asthma can be classified into Type 2 (T2) and non-T2 endotypes based on inflammatory and immune responses. 2 Its heterogeneity, manifested in both clinical phenotypes and molecular endotypes, is influenced by a complex interplay of environmental and genetic factors. 3 These factors, including genetic and epigenetic modulators, are associated with the susceptibility and severity of asthma. 4
Currently, DNA damage response (DDR) has become a promising research area. The vicious cycle between DNA damage and inflammation plays a pivotal role in asthma pathogenesis. Specifically, studies have reported elevated levels of DNA damage in the lung tissue and bronchial epithelial cells of patients with asthma. Oxidative DNA lesions and double-strand breaks (DSBs) are the main threats to DNA integrity in patients with allergic asthma.5,6 Air pollution, specifically exposure to particulate matter (PM) 2.5, increases reactive oxygen species (ROS) levels and amplifies oxidative stress and DDRs. 7 In asthma, excessive ROS production from infiltrating immune cells, particularly eosinophils and neutrophils, along with impaired antioxidant responses, induces oxidative stress, thereby promoting airway inflammation and hyperresponsiveness. 8 Simultaneously, inflammation-induced ROS and reactive nitrogen species not only cause oxidative DNA lesions and DSBs but also impair the function of key proteins in the DNA repair machinery. 9 Additionally, DDR defects can increase the levels of cytosolic double-strand DNA (dsDNA), inducing a cascade of proinflammatory and stress responses, such as cellular senescence, autophagy, apoptosis, and cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING)-mediated inflammation pathways.10–13 Defects in DNA repair pathways enhance the production of proinflammatory cytokines and inflict damage to the bronchial epithelium barriers, thereby substantially contributing to the development of asthma. 14
Furthermore, recent investigations have revealed that several DDR-related molecules play a critical role in driving inflammatory responses and contribute to various inflammatory disorders, particularly asthma. 15 Single nucleotide polymorphisms in the DDR-related genes RAD50 and PARP1 have been linked to asthma susceptibility.16,17 These findings uncovered the potential involvement of DDR mechanisms in modulating asthma pathophysiology. However, the precise mechanisms by which DDR molecules influence asthma progression remain poorly understood.
The present study aimed to identify crucial DDR-related genes as potential diagnostic biomarkers for asthma and explore their upstream and downstream regulatory networks. This was achieved by combining bioinformatics and experimental techniques. Herein, we obtained datasets from the Gene Expression Omnibus (GEO) database to identify and validate DDR-related hub genes. Subsequently, we conducted comprehensive analyses of gene–gene interactions and transcription factor (TF)–microRNA (miRNA)–gene regulatory networks. The methodological flowchart of this study is shown in Figure S1.
Methods
Data acquisition
Five microarray datasets were downloaded from the NCBI GEO database (http://www.ncbi.nlm.nih.gov/geo/) (Table 1). The inclusion criteria were as follows: (a) the species origin should be Homo sapiens; (b) both asthma and control groups included in the datasets; and (c) a clear definition and categorization between the diseased and healthy states. The GSE76262 dataset, derived from induced sputum samples, was selected as the training dataset. Given the nonspecific tissue expression of certain DDR-related genes (https://www.proteinatlas.org/), we utilized datasets of diverse tissues as verification datasets to investigate and compare the tissue-specific expression patterns of asthma-associated DDR-related genes.
Information of GEO datasets.

GEO: Gene Expression Omnibus.
Identification of differentially expressed genes (DEGs)
The normalized data (Series Matrix Files) of the selected datasets were downloaded from the GEO database (Figure S2). To identify DEGs between the asthma and healthy groups, the training set (GSE76262) was initially evaluated. GEO2R, an online analysis tool within the R Project (http://www.ncbi.nlm.nih.gov/geo/geo2r), was utilized to analyze the DEGs between the two groups. Genes were considered as DEGs if they met the following criteria: (a) adjusted p-value/false discovery rate of <0.05 and (b) absolute fold change (|FC|) > 1.5 (|log2 |FC| > 0.5849625). After eliminating the entries in the dataset that lacked corresponding gene symbols or probe IDs, the average expression values of each gene were incorporated for further analyses.
Identification and description of DDR-related DEGs
DDR-related genes were obtained by integrating two databases. The first database comprised 379 genes involved in nine DDR and DNA damage tolerance pathways, including translesion synthesis, homologous recombination (HR), mismatch repair, base excision repair, nonhomologous end joining (NHEJ), chromatin remodeling, nucleotide excision repair (NER), direct repair and telomere repair. The second database contained 299 genes linked to 18 DDR pathways or functions, retrieved from the Human DNA Repair Genes database (https://www.mdanderson.org/documents/Labs/Wood-Laboratory/human-dna-repair-genes.html). By combining these two sources, a final set of 525 DDR-related genes was obtained.
DDR-related DEGs were then identified as the overlapping genes between DEGs and DDR-related genes and were visually presented using a Venn diagram (https://bioinformatics.psb.ugent.be/webtools/Venn/). Volcano plots and expression heatmaps of these DDR-related DEGs were generated using Hiplot (https://hiplot.com.cn/home/index.html) and Sangerbox (http://sangerbox.com/index.html), respectively.
Protein–protein interaction (PPI) analysis and identification of hub DDR-related DEGs
The PPI network of DDR-related DEGs was constructed using the STRING database (https://string-db.org/) and visualized via Cytoscape software (version 3.10.1). By employing the MCODE plug-in in Cytoscape software, we identified the top two gene submodules within the PPI network. Hub genes were indicated in the GSE76262 dataset using the cytoHubba plug-in in Cytoscape software, by applying five algorithms, namely, Degree, MNC, EPC, Closeness, and Radiality. Genes overlapping across these five algorithms were defined as hub DDR-related DEGs.
Enrichment analysis
Using the Metascape platform, hub DDR-related DEGs were analyzed for Gene Ontology (GO) and Kyoto Encyclopedia Gene Genome (KEGG) pathway enrichment. GO analysis, encompassing cellular component (CC), biological process (BP), and molecular function (MF) categories, was performed to elucidate the functional roles of genes. Significantly enriched KEGG and GO pathways were identified based on strict criteria (minimum overlap = 3, p < 0.01, minimum enrichment = 1.5).
Expression and receiver operating characteristics (ROC) analyses
The expression patterns of hub DDR-related DEGs in asthma and healthy groups were analyzed in both the training and verification datasets using SPSS (version 27) and visualized via GraphPad Prisms (version 9). Based on the normality test results, nonparametric tests or independent-sample t tests were employed to assess differences between the two groups within each dataset. Statistical significance was set at p < 0.05.
Subsequently, MedCalc (version 20.100) was utilized to perform ROC analysis to evaluate area under the curve (AUC) values and predictive abilities across all datasets. Genes exhibiting moderate-to-strong predictive values (AUC > 0.7) in the training dataset and at least one verification dataset were identified as diagnostic biomarkers for asthma, with statistical significance set at p < 0.05.
Construction of TF–target gene and miRNA–target gene networks
To explore the regulatory relationships between TFs, miRNAs, and key diagnostic DDR-related genes in asthma, TF–target gene and miRNA–target gene networks were constructed using the NetworkAnalyst platform (https://www.networkanalyst.ca). TF–target gene interactions were predicted using the JASPAR database (https://jaspar.genereg.net), in conjunction with TF-related Disease Ontology (DO) terms from the TRRUST database (https://www.grnpedia.org/trrust/). Disease–miRNA associations were sourced from the HMDD.v4.0 database (www.cuilab.cn/hmdd), and miRNA–TF regulatory relationships were searched from the TransmiR v3.0 database (http://www.cuilab.cn/transmir). Additionally, miRNA functional enrichment analysis was performed using the DIANA-miRPath v4.0 database (http://www.microrna.gr/miRPathv4). The TarBase v.8 database (https://dianalab.e-ce.uth.gr/html/diana/web/index.php?r=tarbasev8) was utilized to identify interactions between key diagnostic DDR-related genes and miRNAs. The resulting regulatory networks, integrating DDR-related genes, TFs, and miRNAs, were constructed and visualized using Cytoscape.
Correlation and expression analysis
The spearman correlation between key diagnostic DDR-related genes and inflammatory genes or TFs were assessed using SPSS (version 27) and presented via Hiplot, with significant correlation defined by a p-value of <0.05 and correlation coefficient (r) of ≥0.3. In the five datasets, asthma samples were divided into normal and lower/higher expression groups based on the median expression levels of key diagnostic DDR-related genes. Nonparametric tests or independent-sample t tests were used for group comparisons depending on normality test results, with p < 0.05 considered statistically significant.
RT–qPCR analysis of potential key DDR-related genes
This was a case–control study strictly adhering to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guideline. 18 All participants demonstrated strong comprehension and cooperation abilities and were neither pregnant nor lactating. The case group comprised adult patients with asthma who had no respiratory tract infections within the past 4 weeks or serious diseases affecting other systems. The control group consisted of adults without respiratory tract infections within the past 4 weeks and no history of respiratory diseases, allergic conditions, or other serious systemic disorders. From June 2022 to February 2024, a total of 12 healthy controls and 35 patients with asthma were recruited from the Allergy and Clinical Immunology Department of the First Affiliated Hospital of Guangzhou Medical University. The diagnosis of asthma and the determination of endotypes (T2 and non-T2 asthma) were in line with the Global Initiative for Asthma guidelines. Whole blood samples were collected to isolate peripheral blood mononuclear cells (PBMCs). For patients with asthma undergoing bronchoscopy, bronchial mucosal tissues were obtained to prepare primary human bronchial epithelial cells (pHBECs) following established protocols.
The study protocol was approved by the Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University (Approval No. 2022075) on 18 May 2022 in Guangdong, China. This study complied with the ethical principles for medical research involving human subjects, as outlined in the Declaration of Helsinki (1975) and its subsequent amendments, including the 2013 revision. All participants or their legal guardians signed the informed consent forms before the collection of blood samples and bronchial mucosal tissues.
Following the manufacturer’s instructions, total RNA was extracted from PBMCs or pHBECs using Trizol Reagent (Ambion, USA, Cat#15596026). The RNA was then reverse transcribed into cDNA (Vazyme, China, Cat#R323) and used for RT–qPCR amplification (Vazyme, China, Cat#Q711). In the experiment, both technical replicates and biological replicates were performed three times. The primer sequences for potential key DDR-related genes are listed in Table S1. The mRNA expression levels were normalized to that of GAPDH, which served as an internal control, and quantitatively analyzed using the 2−ΔΔCT method. The thermocycling protocol for this study began with an initial denaturation step at 95°C for 2 min. Subsequently, 40 amplification cycles were performed. Each cycle consisted of a denaturation phase at 95°C for 10 s and an annealing step at 60°C for 30 s. After the amplification cycles, a melt-curve analysis was conducted, spanning from 65°C to 95°C with a temperature increment of 0.5°C every 5 s.
Results
Identification of DDR-related DEGs and PPI network analysis
In the GSE76262 dataset, 1560 DEGs were identified between asthma and healthy groups. Among these DEGs, 28 were recognized as DDR-related DEGs (Figure 1(a), Table S2). Specifically, 24 of these DDR-related DEGs were downregulated in asthmatic samples. In contrast, four genes, namely, NBN, GADD45B, PER1, and BLM, were upregulated (Figure 1(b), Figure S3A).
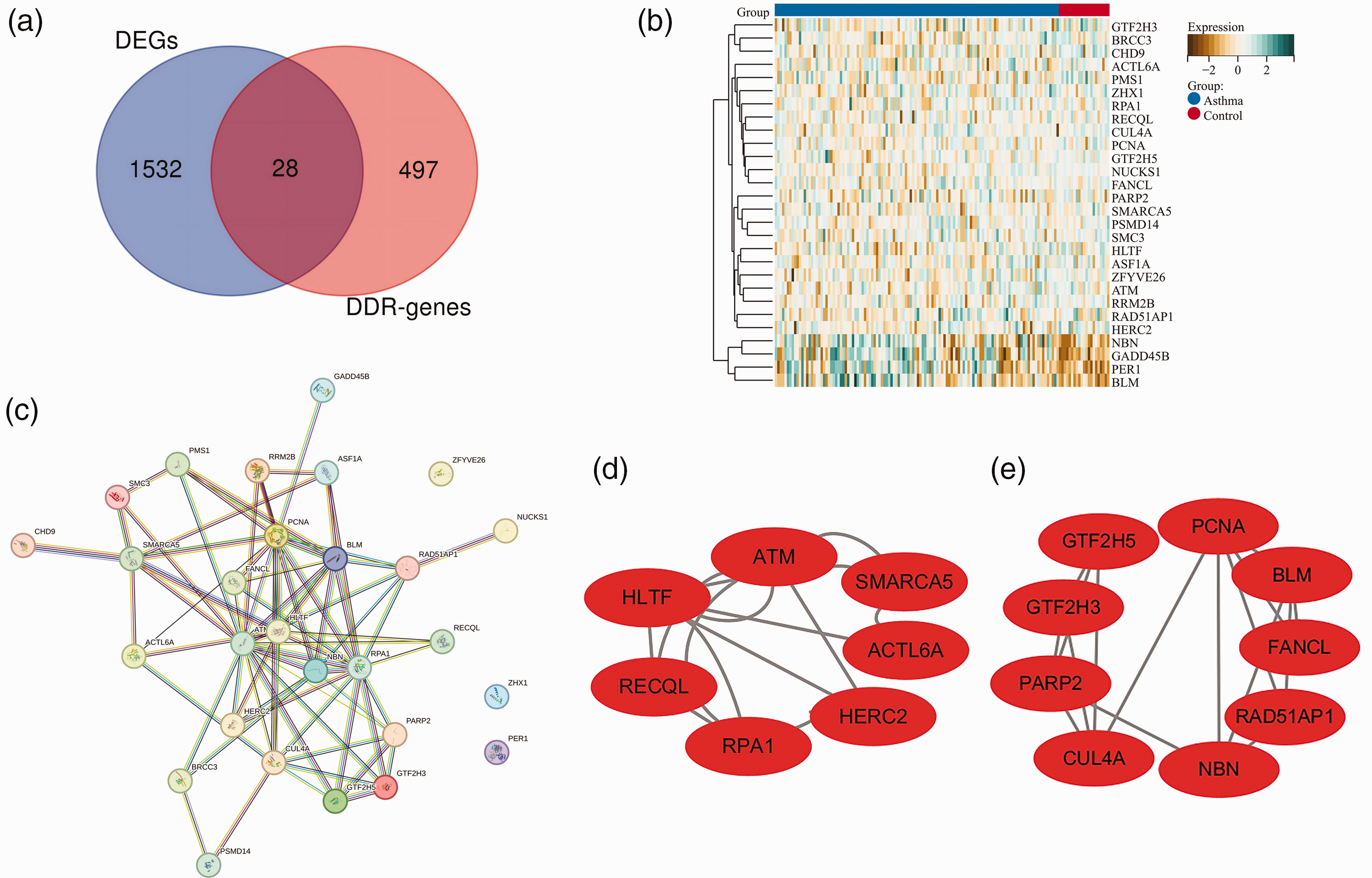
Identification and analysis of DDR-related DEGs in the GSE76262 dataset. (a) A Venn diagram presenting the overlap between DEGs in asthma and those related to DDR. (b) A heatmap illustrating the expression patterns of 28 DDR-related DEGs, with green and brown colors representing up- and downregulation, respectively. (c) A PPI network revealing protein association among 28 DDR-related DEGs. Light blue and purple edges represent known interactions, while dark blue, green, and red edges indicate predicted interactions. (d and e) Visualization of the top two submodules, comprising 25 interacting DDR-related DEGs, using Cytoscape software. DDR: DNA damage response; DEG: differentially expressed gene; PPI: protein–protein interaction.
A PPI network analysis was then performed. The results revealed that 25 of the 28 DDR-related DEGs interacted with each other, whereas ZFYVE26, ZHX1, and PER1 did not participate in these interactions (Figure 1(c)). Subsequently, the top two submodules with the highest clustering scores were identified, which contained 16 key genes (Figure 1(d) and (e)). These 16 genes were further subjected to KEGG and GO enrichment analyses. The KEGG enrichment analysis revealed that these genes were significantly enriched in several pathways, including HR, NER, cell cycle, and ubiquitin-mediated proteolysis (Figure S3B). Furthermore, GO enrichment analyses of BP, MF, and CC indicated significant enrichment in DNA damage recognition and processing pathways (Figure S3C–E).
Hub genes recognition and validation analysis
The top 10 genes were identified using the cytoHubba plug-in via five algorithms (Figure 2(a), Figure S4). Among these, nine overlapping genes were determined as hub DDR-related DEGs (Figure 2(b)). The expression of these hub DDR-related DEGs was analyzed in the GSE76262 dataset. The results revealed that compared with healthy individuals, ATM, RPA1, PCNA, HLTF, CUL4A, RAD51AP1, and PARP2 were significantly downregulated in patients with asthma, whereas BLM and NBN exhibited higher expression levels (Figure 2(c)). To validate the reliability of the training dataset, the mRNA expression profiles of these hub DDR-related DEGs were examined in verification datasets. As shown in Figure S5, all hub DDR-related DEGs displayed significant differences (ATM: p = 0.001 in GSE74986 and p < 0.001 in GSE69683; RPA1: p = 0.019 in GSE69683; PCNA: p < 0.001 in GSE74986; HLTF: p = 0.011 in GSE137268; CUL4A: p < 0.001 in GSE74986 and GSE69683; RAD51AP1: p < 0.001 in GSE74986; PARP2: p = 0.006 in GSE137268 and p = 0.002 in GSE74986; BLM: p = 0.004 in GSE69683; NBN: p = 0.001 in GSE137268, p < 0.001 in GSE74986, and p = 0.028 in GSE43696) and consistent trends in at least one verification dataset, qualifying them as verified hub genes for further analysis. Notably, the expression of NBN mRNA was upregulated in asthma samples from GSE76262 (induced sputum), GSE137268 (induced sputum), and GSE43696 (bronchial epithelial cells) datasets, but downregulated in the GSE74986 (bronchoalveolar lavage cells) dataset. Notably, none of these genes were consistently verified across all datasets, probably due to variations in sample sources and limited sample sizes.

Identification and validation of DDR-related hub genes in the GSE76262 dataset. (a) A Venn diagram presenting the overlapping hub genes identified via five clustering algorithms. (b) The list of top 10 DDR-related DEGs identified via five clustering algorithms. (c) Expression level of the hub genes between asthma (red) and control (blue) samples in the training dataset. Genes with significant expression changes are presented in bold format. Significance levels are indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001. (d and e) The ROC curves of 6 DDR-related hub genes with AUC values of >0.7 were analyzed separately (d) or combined (e) in the GSE76262 dataset. Significance levels are indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001. DDR: DNA damage response; DEG: differentially expressed gene; ROC: receiver operating characteristic; AUC: area under the curve.
The ROC curves of nine hub genes are presented in Table S3. In the GSE76262 dataset, seven genes exhibited AUC values exceeding 0.7. Notably, six of these genes, namely, PARP2, CUL4A, ATM, NBN, PCNA, and HLTF, had AUC values greater than 0.7 in one or more verification datasets. Thus, they were identified as potential key diagnostic DDR-related genes for asthma (Figure 2(d), Figure S6A–D). The combination of these six genes exhibited enhanced predictive power across different datasets, with AUC values reaching 0.821 in GSE76262, 0.770 in GSE137268, 0.941 in GSE74986, 0.663 in GSE43696, and 0.706 in GSE6983 (Figure 2(e), Figure S6E–H). These findings indicate that PARP2, CUL4A, ATM, NBN, PCNA, and HLTF can serve as potential diagnostic biomarkers for asthma.
Identification of downstream effectors and key diagnostic DDR-related genes in asthma via expression analysis
The asthma samples from the GSE76262 dataset and the four verification datasets were categorized into normal and lower/higher expression groups based on the median expression levels of the key diagnostic DDR-related genes. Subsequently, correlation analysis (Figure 3, Table S4) and expression profiling (Table S5) were performed to explore the relationships between key DDR-related genes and inflammation-related genes in asthma. These inflammation-related genes include those involved in oxidative stress, autophagy, apoptosis, senescence-associated secretory phenotype (SASP), and cGAS–STING signaling as well as T2 and non-T2 cytokines.

Correlation analysis of key DDR- and inflammation-related genes in asthma samples. This correlation was analyzed in asthma samples across five datasets: GSE76262 (a), GSE137268 (b), GSE74986 (c), GSE43696 (d), and GSE69683 (e). Circles without “X” inside indicate significant correlations, with red circles representing positive correlations and purple circles representing negative correlations. DDR: DNA damage response.
Six key DDR-related genes exhibited significant negative correlations with T2 genes (IL33 and CSF2) in various samples of patients with asthma, especially in bronchoalveolar lavage fluid (BALF) and sputum, reflecting an increase in T2 airway inflammation. Specifically, five of these DDR genes (ATM, PCNA, PARP2, CUL4A, and HLTF) consistently displayed negative correlations with genes involved in the cGAS–STING pathway (TBK1, NFKB1, TGFB1) and positive correlations with autophagy-related genes (BECN1, ATG5, ATG7) (Table S4). These results suggest that the downregulation of DDR genes may be accompanied by activation of the cGAS–STING pathway and inhibition of the autophagy pathway (Table S4).
Expression analysis further validated these findings. In the groups with lower expression of ATM, PCNA, HLTF, CUL4A, and PARP2, the expression levels of T2 and cGAS–STING genes were significantly higher than those in the normal expression groups, whereas the expression levels of autophagy-related genes were significantly lower (Table S5). Conversely, higher NBN expression resulted in the upregulation of multiple inflammation-related genes, such as TBK1, NFKB1, IFNγ, IL1A, and CXCL1, but led to the downregulation of T2 genes. Collectively, these results suggest that the downregulation of key diagnostic DDR-related genes in patients with asthma can exacerbate T2/non-T2 inflammation through the modulation of cGAS–STING, autophagy, and apoptosis pathways.
Identification of upstream regulators of key diagnostic DDR-related genes in asthma through TF–target gene and miRNA–target gene interaction network analysis
To elucidate the upstream regulators of six key diagnostic DDR-related genes in asthma, we conducted comprehensive analyses of TF–target gene and miRNA–target gene interaction networks. We identified 34 TFs associated with the six DDR-related genes, among which 15 have been linked to asthma pathogenesis (Figure 4(a)). These identified TFs were used as the foundation for further analysis. Correlation, expression, and enrichment analyses of these TFs and the key diagnostic DDR-related genes were conducted in asthma groups from five datasets (Table S6, Figure S7A–B). Notably, in various samples such as sputum and BALF, PCNA, ATM, PARP2, HLTF, and CUL4A showed significant negative correlations with TFs that are upregulated in asthma. In contrast, these genes showed significant positive correlations with TFs that are downregulated in asthma. However, the correlation pattern between NBN and TFs was opposite.

Upstream regulators of key diagnostic DDR-related genes in asthma. (a and b) Predicted interaction network between TFs and DDR-related genes (a) as well as miRNAs and DDR-related genes (b). Red spots indicate DDR-related genes, whereas blue and purple rectangles represent TFs and miRNAs, respectively. (c–h) The TF–miRNA–target gene network analysis of ATM (c), PCNA (d), HLTF (e), CUL4A (f), PARP2 (g), and NBN (h). Red ovals represent target DDR-related genes, purple rectangles represent miRNAs, and blue diamonds represent TFs. Red lines denote positive regulation of target genes, whereas green lines represent negative regulation. Black dotted lines indicate both positive and negative regulation.
The miRNA–target gene interaction network analysis revealed that 46 miRNAs were associated with NBN, 60 with PCNA, 64 with ATM, 8 with PARP2, 33 with HLTF, and 38 with CUL4A (Figure 4(b)). Among the top 15 miRNAs linked to at least three of these key DDR-related genes, hsa-miR-103a-3p, hsa-miR-92a-3p, and hsa-miR-26a-5p have been reported to promote inflammatory responses in asthma (Table S7). As presented in Figure S7C–D and Table S8, these three miRNAs may regulate the expression of key diagnostic DDR-related genes through both DDR and inflammation pathways.
To gain a deeper understanding of the upstream regulators of DDR-related genes in asthma, we conducted a comprehensive regulatory network analysis encompassing three miRNAs (hsa-miR-103a-3p, hsa-miR-26a-5p, and hsa-miR-92a-3p), 11 TFs, and six target genes (Figure 4(c)–(h)). Our results demonstrated that these miRNAs can directly or indirectly downregulate the expression of key diagnostic DDR-related genes (ATM, PCNA, PARP2, HLTF, and CUL4A) by upregulating four specific TFs, namely, GATA2, E2F1, NFKB1, and SRF. Additionally, we observed that the expression of these target DDR-related genes (ATM, PCNA, HLTF, CUL4A, and PARP2) was directly downregulated by various proinflammatory TFs, such as GATA2, REL, E2F1, NFKB1, and SRF, but was upregulated by anti-inflammatory TFs such as PPARG, CREB1, and DDR-related TFs (e.g. YY1, TP53, and SP1).
Experimental validation of potential key DDR-related genes
The expression of potential key DDR-related genes was experimentally validated in PBMCs derived from healthy controls and patients with asthma. As shown in Figure 5, the expression of ATM in patients with asthma was significantly lower than that in healthy individuals. In contrast, there were no significant variations in the expression of PCNA and NBN between the two groups. PARP2 and CUL4A exhibited a tendency toward downregulation in asthma, consistent with the expression patterns observed in the GSE69683 dataset (Figure S5).

Experimental validation of key DDR-related genes in PBMCs. The expression levels of six key DDR-related genes—ATM (a), PCNA (b), HLTF (c), CUL4A (d), PARP2 (e), and NBN (f)—were detected in 12 healthy individuals and 35 patients with asthma (including 28 individuals with T2 asthma and 7 with non-T2 asthma). Significance levels are indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001. DDR: DNA damage response; PBMCs: peripheral blood mononuclear cells.
Our preliminary analysis suggests that the expression of these genes vary across different asthma endotypes and individual samples. Specifically, the observed differences in the expression levels of ATM, PARP2, and CUL4A between patients with asthma and healthy controls in PBMCs were primarily attributed to the contrast between T2 asthma and healthy controls, rather than non-T2 asthma. In PBMCs, the expression levels of ATM and PARP2 were significantly lower in T2 asthma than in non-T2 asthma (Figure 5). This finding was successfully corroborated in pHBECs (Figure S8). This disparity may be attributed to the complex endotypes of asthma. Furthermore, these results are in line with the correlation and expression analyses presented in Table S4 and Table S5, which highlight a negative correlation between DDR genes and T2 cytokines.
The expression of HLTF was significantly downregulated in patients with asthma compared with that in healthy controls in PBMCs. A same tendency was also found in the GSE69683 dataset (Figure S5). Notably, this difference was not influenced by the T2 or non-T2 endotypes as HLTF expression was significantly lower in T2 asthma than in non-T2 asthma in both PBMCs and pHBECs (Figure S8). The underlying reasons for this discrepancy require further investigation and discussion.
Discussion
The current study intended to clarify the role of DDR-related genes in asthma pathogenesis. By utilizing bioinformatics approaches, we first identified nine hub DDR-related DEGs in asthma, which are primarily implicated in HR and NER pathways. Notably, six hub genes, namely PCNA, HLTF, NBN, ATM, PARP2, and CUL4A, were identified as potential diagnostic DDR-related genes for asthma. Our findings further revealed that the downregulation of certain key genes in asthma is associated with the increased expression of T2-, cGAS–STING-, and SASP-related genes as well as the reduced expression of autophagy-related genes. Our findings suggest a complex interplay between DDR pathways and other asthma-associated molecular processes. Moreover, a comprehensive miRNA–TF–target gene regulatory network analysis illuminated the regulatory roles of three miRNAs (hsa-miR-103a-3p, hsa-miR-26a-5p, and hsa-miR-92a-3p) and 11 TFs (REL, RELA, E2F1, TP53, YY1, CREB1, PPARG, NFKB1, GATA2, SRF, and SP1) in modulating the expression of the six key diagnostic DDR-related genes. Finally, experimental studies validated the downregulated expression of ATM and HLTF in patients with asthma and newly discovered variations in the expression of these genes across different asthma endotypes.
Downregulation of DDR leads to elevated DNA damage in asthma
The six key diagnostic genes for asthma identified in our study play crucial roles in oxidative DNA damage repair and DSB repair pathways. ATM, NBN, and PARP2 act as damage sensors to initiate DNA repair, whereas PCNA, HLTF, and CUL4A are involved in the downstream repair processes. Both in vivo and in vitro studies have demonstrated that exposure to environmental hazards such as house dust mite or PM 2.5 can cause oxidative DNA damage and DSBs in lung tissue and bronchial epithelial cells, potentially contributing to asthma pathogenesis.5,6,14 In patients with asthma, excessive production of ROS results in increased levels of oxidative DNA damage in sputum, serum, urine, and BALF samples, with 8-oxo-7,8-dihydroguanine being the main product. 19 The replication of single-strand breaks, mainly caused by oxidative ROS attack, is a physiological source of DSBs. 20 Additionally, physiological DSBs can occur during V(D)J recombination and immunoglobulin heavy chain class switch recombination, which are exclusively repaired through the NHEJ pathway and presumably through the end joining pathway. 21 Therefore, immune cells such as T and B cells may exhibit high sensitivity to DSBs, as evidenced by the increased DNA damage levels in lymphocytes of children with asthma. 22 In addition, pediatric patients with asthma showed a higher frequency of spontaneous sister chromatid exchanges, indicating an increase in DSBs and a reduction in HR repair activities in asthma. 23
In summary, the reduced expression of key DDR genes in asthma impairs the repair of DNA damage. Consequently, this leads to the accumulation of unrepaired DNA and the initiation of DNA damage-driven inflammatory processes.24,25
DNA damage-induced inflammatory responses in asthma pathogenesis
The interplay between DDR and immune responses has been increasingly recognized as a contributing factor to inflammatory disorders, particularly asthma.26,27 DDR molecules responsible for recognizing and processing DNA damage are crucial for initiating and maintaining the production of inflammatory cytokines. 25 DNA damage-induced cytosolic dsDNA, micronuclei, and the accumulation of inflammasome can activate the cGAS–STING and nuclear factor kappa light chain enhancer of activated B cells (NF-κB) signaling pathways, thereby promoting the secretion of proinflammatory cytokines. 24 Furthermore, the DDR machinery is involved in autophagy and apoptosis signaling, both of which are essential for airway remodeling and T2 inflammation in asthma.28,29
Accumulating evidence indicates that PARP1 and poly ADP-ribosylation (PARylation) play a crucial role in asthma, as PARP1 regulates T2 responses through STAT-6 cascade. 30 Additionally, PARP1 has been implicated in promoting Type 17 (T17) inflammation in T cells and keratinocytes, suggesting a role in inflammatory bowel diseases and psoriasis.31,32 In our current study, we found that the mRNA expression of PARP2 was significantly reduced in induced sputum, BALF, and peripheral blood samples from patients with asthma. PARP2 is a member of the PARP superfamily enzymes that generate PARylation as an immediate response to oxidative DNA damage and DSBs. 33 Lower expression levels of PARP2 were associated with upregulated T2 responses, suggesting that reduced PARP2 activity can aggravate T2 inflammation in asthma. However, the specific mechanisms and implications of PARP2-mediated PARylation in the pathogenesis of asthma remain to be further investigated. Moreover, it has been reported that PARP2 positively regulates autophagic degradation in transformed mammalian cells, and its absence hinders the autophagy process. 34 Our results revealed that the reduced expression of the autophagic factor ATG5 in cells with lower PARP2 levels suggests a potential involvement of PARP-dependent autophagy in asthma pathogenesis.
Previous studies have shown that both ATM and the MRE11-RAD50-NBN (MRN) complex, with RAD50 being the primary component, can promote PM-induced Type 1 (T1) airway inflammation in mice, possibly due to dysregulated DDR mechanisms.14,35 ATM and NBN are also necessary in SASP induced by persistent DNA damage, which leads to an increase in IL-6 secretion. 25 Surprisingly, we found that the expression of ATM was decreased, while the levels of NBN were increased in patients with asthma. Additionally, the downregulation of ATM was accomplished by elevated T2 cytokines expression, whereas the upregulation of NBN triggered the activation of T1 and T17 signaling. We also confirmed that in patients with asthma, a lower expression of NBN in the BALF sample was accompanied with higher levels of NFKB1, TNF, TGFB1, and CXCL1. Previous studies have indicated that MRE11 and RAD50, two crucial components of the MRN complex, are essential for the interferon response induced by cytosolic dsDNA-STING, while NBN is not necessary. 36 However, NBN, in coordination with ATM and carboxy-terminal binding protein 1 interacting protein (CtIP), can prevent cGAS from binding to micronuclei and subsequent activation of inflammatory signaling. Cells lacking NBN and CtIP not only recruit cGAS to micronuclear DNA but also activate it, resulting in augmented immune signaling and cellular senescence. Therefore, NBN acts as an upstream regulatory factor that prevents cGAS binding to micronuclear DNA by promoting CtIP-mediated end resection. 37 Nevertheless, further research is needed to elucidate how NBN-mediated suppression of cGAS signaling affects asthma progression.
PCNA, a proliferation-associated protein, has been reported to be upregulated in the epithelium of corticosteroid-dependent asthma. Moreover, its expression level shows a positive correlation with the epithelium thickness. 38 Concurrently, HLTF acts as a ubiquitin ligase, facilitating the ubiquitination of PCNA. This ubiquitination process promotes error-free post-replication repair and restrains cellular proliferation. 39 In our current study, we observed a downregulation of both PCNA and HLTF mRNA expression in the bronchial epithelial cells of patients with T2 asthma. This finding was further confirmed in induced sputum samples, where a significant reduction in the mRNA level of PCNA and HLTF mRNA was accompanied by an upregulation of T2 inflammation signals. Notably, in patients with asthma who had low expression of PCNA and HLTF, the expression patterns of cell cycle-related CDKN1A and autophagy-related ATG5 show opposite trends. Specifically, the upregulation of CDKN1A in the PCNA-low asthma group was hypothesized to hinder cell cycle progression, ultimately leading to an increase in cell apoptosis. In contrast, the upregulation of ATG5 in HLTF-low group is likely to promote autophagy in asthma.
CUL4A, which belongs to E3 ubiquitin ligase family, has a wide spectrum of substrates. It is hypothesized to mitigate oxidative stress-induced inflammation and apoptosis via the NF-κB pathway. 40 Consistently, in patients with asthma, a reduction in CUL4A expression was associated with an upregulation of NF-κB and T2 genes, indicating a potential regulatory role of CUL4A in the pathogenesis of asthma.
Potential TFs and miRNAs regulate DDR expression in asthma
To elucidate the potential regulatory mechanisms governing key diagnostic DDR-related genes, we conducted a comprehensive analysis of the miRNA–TF–target gene regulatory network. In this study, we identified three miRNAs—hsa-miR-103a-3p, hsa-miR-92a-3p, and hsa-miR-26a-5p—which are known to activate inflammatory responses in asthma. Specifically, mast cell-derived hsa-miR-103a-3p exacerbated eosinophilic allergic inflammation through T2 innate lymphoid cell activation. 41 hsa-miR-92a-3p regulates the expression levels of SLC14A1, SNCA, TNS1, KAT2B, and PARP1 in patients with combined allergic rhinitis and asthma. 42 It is also implicated in cigarette smoke extract-induced hyperinflammation in chronic obstructive pulmonary disease and lipopolysaccharide-induced pulmonary inflammation and injury through the regulation of the PTEN/AKT/NF-κB and apoptotic signaling pathways. 43 Furthermore, hsa-miR-26a-5p, which is upregulated in the lung tissue and airway smooth muscle cells in asthmatic mice or horses, may contribute to asthma progression mainly through the regulation of inflammation and cell proliferation. 44
These miRNAs exhibited a negative correlation with the key diagnostic DDR-related genes. They may either directly downregulate the expression of target DDR genes or indirectly achieve this through the upregulation of TFs associated with inflammation or DDR. Among these TFs, GATA2, REL, E2F1, and NFKB1 emerged as proinflammatory TFs involved in asthma, whereas PPARG is an anti-inflammatory TF that suppresses the synthesis and release of immune-modulating cytokines and eosinophil function.45,46 Notably, in individuals with asthma, PPARG expression is associated with airway inflammatory and remodeling responses. 47 YY1, TP53, and SP1 are TFs related to DDR. YY1 specifically binds to the T2 locus control region and the entire T2 cytokine locus in a T2-specific manner, inducing T2 cytokine expression. In asthmatic mice, CD4-specific knockdown of YY1 caused a substantial decrease in T2 cytokine expression, repression of chromatin remodeling, reduction of intrachromosomal interactions, and enhanced resistance. 48 Gene polymorphisms in TP53 are linked to asthma susceptibility, and the CpG island in TP53 is related to the severity of late-onset asthma, indicating its role in the induction and progression of asthma. 49 In contrast, SP1 serves as a positive transcriptional regulator of T-Box expressed in T cells (T-BET) and is a crucial factor in T helper cell differentiation and airway immunopathology in asthma. 50 Targeting SP1 could aid in preventing or treating diseases associated with aberrant T-BET expression. 51
Collectively, our findings identified six DDR genes as potential diagnostic biomarkers for asthma. We have also provided preliminary evidence regarding a vicious cycle of inflammation-induced DNA damage and damage-induced inflammation, which may exacerbate asthma progression. Moreover, our study contributes to clinical practice by enhancing the theoretical understanding of DDR-related biomarkers and guiding future research on underlying mechanisms. This may lead to novel therapeutic approaches for patients with asthma. However, the current research has several limitations. First, the transcriptomic and clinical data were obtained from public databases, with varying sample types and sizes, which may affect the accuracy of the results. Second, although in vitro validations revealed distinct expression patterns of DDR molecules across different asthma endotypes, multi-center, larger-sample analyses are needed to verify our findings due to the limited sample sizes of both public datasets and our validation cohort. Moreover, further studies are required to elucidate the specific roles of key diagnostic DDR-related genes in asthma, especially the causal relationship between inflammation and DDR.
Conclusion
Six DDR genes related to oxidative and double-strand DNA damage repair were first identified as potential key diagnostic genes in asthma through bioinformatics and experimental approaches. The miRNA–TF–target DDR-related gene network and correlation analysis revealed a vicious cycle of inflammation and DNA damage in asthma pathogenesis. This finding provides potential therapeutic avenues targeting DNA damage and DDR in asthma treatment.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605251332204 - Supplemental material for Identification of hub genes related to DNA damage response in asthma via combinative bioinformatics strategy
Supplemental material, sj-pdf-1-imr-10.1177_03000605251332204 for Identification of hub genes related to DNA damage response in asthma via combinative bioinformatics strategy by Li He, Fangmei Lin, Yawen Zhou, Meihua Dong, Mingfang Deng, Jing Li and Nan Jia in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605251332204 - Supplemental material for Identification of hub genes related to DNA damage response in asthma via combinative bioinformatics strategy
Supplemental material, sj-pdf-2-imr-10.1177_03000605251332204 for Identification of hub genes related to DNA damage response in asthma via combinative bioinformatics strategy by Li He, Fangmei Lin, Yawen Zhou, Meihua Dong, Mingfang Deng, Jing Li and Nan Jia in Journal of International Medical Research
Supplemental Material
sj-pdf-3-imr-10.1177_03000605251332204 - Supplemental material for Identification of hub genes related to DNA damage response in asthma via combinative bioinformatics strategy
Supplemental material, sj-pdf-3-imr-10.1177_03000605251332204 for Identification of hub genes related to DNA damage response in asthma via combinative bioinformatics strategy by Li He, Fangmei Lin, Yawen Zhou, Meihua Dong, Mingfang Deng, Jing Li and Nan Jia in Journal of International Medical Research
Footnotes
Acknowledgements
Not applicable.
Authors’ contributions
N.J. and L.H. designed the study; L.H., MH.D., and Y.Z. performed data acquisition; L.H. conducted bioinformatics and statistical analysis; L.H., F.L., and MF.D. performed qPCR experiments with in-house samples; N.J., L.H., and J.L. coordinated the study; L.H. and N.J. wrote the manuscript; and all authors commented on the manuscript.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics consideration
The study protocol received approval from the Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University (No. 2022075), in accordance with the Declaration of Helsinki. Informed consent forms were obtained from all participants or their legal guardians.
Funding
This work was supported through the grants of National Natural Science Foundation of China (82300029), Guangdong Basic and Applied Basic Research Foundation (2020A1515111181), and Science and Technology Projects of Guangzhou (202102010361) provided to N.J. as well as grants of National Natural Science Foundation of China (82161138020, U1801286) and Zhongnanshan Medical Foundation of Guangdong Province (ZNSA-2020013) provided to J.L. We appreciate the Biobank for Respiratory Diseases in the National Clinical Research Center for Respiratory Disease (BRD-NCRCRD, Guangzhou, Southern China) for providing sample preservation services for this work.
List of abbreviations
AUC: area under the curve; BALF: bronchoalveolar lavage fluid; BP: biological process; CC: cellular component; cGAS: cyclic GMP–AMP synthase; CtIP: carboxy-terminal binding protein 1 interacting protein; DDR: DNA damage response; DEGs: differentially expressed genes; DO: Disease Ontology; DSB: double-strand break; dsDNA: double-strand DNA; FC: fold change; GEO: Gene Expression Omnibus; GO: Gene Ontology; HR: homologous recombination; KEGG: Kyoto Encyclopedia Gene Genome; MF: molecular function; miRNA: microRNA; MRN: MRE11–RAD50–NBN; NER: nucleotide excision repair; NF-κB: nuclear factor kappa light chain enhancer of activated B cells; NHEJ: nonhomologous end joining; PARylation: poly ADP-ribosylation; PBMCs: peripheral blood mononuclear cells; pHBEC: primary human bronchial epithelial cells; PM 2.5: particulate matter 2.5; PPI: protein–protein interaction; ROC: receiver operating characteristic; ROS: reactive oxygen species; RT–qPCR: reverse transcription polymerase chain reaction; SASP: senescence-associated secretory phenotype; STING: stimulator of interferon genes; STROBE: Strengthening the Reporting of Observational Studies in Epidemiology; T-BET: T-Box expressed in T cells; TF: transcription factor; T1: Type 1; T2: Type 2; T17: Type 17.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.