
Abstract
Objective
To characterize the gut microbial composition of patients with optic neuritis (ON) or myasthenia gravis (MG).
Methods
Stool samples were collected from 45 patients with ON, 13 patients with MG, and 20 healthy controls. Microbial genomic DNA was extracted, and the V3–V4 regions of bacterial 16S rRNA genes were amplified and sequenced. Bioinformatic analyses was performed to compare the alpha-diversity, beta-diversity, taxonomic assignments, and bacterial richness of the groups. Differences in the abundances of microbial taxa were identified using linear discriminant analysis effect size (LEfSe) and analysis of variance.
Results
Beta-diversity analysis showed distinct clustering of patient samples from the healthy controls. At the phylum and genus levels, Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria predominated, but their proportions varied between groups. LEfSe analysis identified microbial taxa that were associated with each group. The patients showed lower abundances of certain intestinal probiotics, including Bifidobacterium, Bacteroides, and Roseburia, than the controls. No significant differences were found between the disease subgroups. The Carnobacteriaceae family was significantly less abundant in the ON than in the MG group.
Conclusion
We have identified significant alterations in the gut microbiota of patients with ON or MG, and importantly, a notable reduction in intestinal probiotics.
Keywords
Introduction
Optic neuritis (ON) and myasthenia gravis (MG) are neuroimmune disorders that affect the optic nerve and the neuromuscular junction, respectively, and have distinct clinical manifestations. ON is an inflammation of the optic nerve, which can be associated with inflammatory disease of the central nervous system (CNS) or can occur in isolation. Patients often present with partial or complete acute vision loss and eye pain within a few days of onset. 1 ON can be categorized into multiple subtypes according to its association with specific underlying conditions. 2 Multiple sclerosis-associated optic neuritis (MS-ON) is linked to multiple sclerosis (MS) and typically presents with features of demyelination. This manifestation is also referred to as ‘typical ON (T-ON)’ or ‘idiopathic demyelinating optic neuritis (IDON),’ thereby differentiating it from atypical forms of ON, such as inflammatory optic neuropathy (ION), which develops secondary to systemic connective tissue diseases, and neuromyelitis optica spectrum disorder-associated optic neuritis (NMOSD-ON).3,4 NMOSD-ON may exhibit distinct clinical and radiologic characteristics to T-ON. This subtype is often associated with the presence of aquaporin-4 antibodies (AQP4-IgG). 5 In some cases of ON, and in particular those not associated with MS or NMOSD, antibodies targeting myelin oligodendrocyte glycoprotein (MOG-IgG) may be present. 6 ION is used for cases in which the ON is related to systemic inflammatory conditions other than MS and NMOSD. 1 MG is a neuroimmune disorder that is characterized by immune-mediated disruption of the neuromuscular junction, and patients often present with ptosis and eye movement abnormalities, owing to weakness of the eye muscles, and the ocular type (OMG) is the mildest clinical subtype.
In recent years, significant alterations in gut microbial composition have been reported to characterize neuroinflammatory disorders.7,8 The composition of the microbiota in patients with MS has been explored in a number of studies, and the composition of the microbiota in patients with MS and healthy individuals has been shown to differ. 9 In addition, the gut microbiota and its metabolites plays a significant role in the pathophysiology of NMOSD, and may serve as biomarkers for disease onset and progression and as targets for disease-modifying therapy. 10 Interestingly, previous studies have shown lower proportions of Verrucomicrobiaceae and Bifidobacteriaceae in patients with MG. 11 Tan et al. showed that the various subtypes of MG are associated with differences in gut microbial composition. 12 Because targeting of gut microbial dysbiosis represents a potential therapeutic strategy for neurological disorders, 13 a better understanding of the intricate relationship between the gut microbial composition and neurological disorders affecting the visual system could pave the way for the development of innovative diagnostic biomarkers and personalized therapeutic approaches.
Although the roles of the gut microbiota in neuroinflammatory and neuroimmune disorders is increasingly recognized, the findings made with respect to the various conditions have been inconsistent. ON and MG, despite affecting different anatomical sites (ON affects the optic nerve and MG affects the neuromuscular junction), are both immune-mediated neurologic disorders, and therefore there is a rationale for a comparative analysis to be performed. The study of ON and MG together could be used to identify both shared and unique gut microbial characteristics of immune-mediated neurologic diseases. In addition, such a comparison would permit the use of ON and MG as complementary models to facilitate understanding of the tissue- or mechanism-specific microbial patterns characterizing immune-mediated conditions. Notably, no previous studies have considered these two diseases together, and therefore this approach could provide new insight into the similarities and differences in the gut microbiota of immune-mediated neurological disorders, potentially informing the development of microbiota-related biomarkers and personalized therapies.
16S rRNA sequencing is a widely used molecular technique in research into the microbiota. It involves the amplification and sequencing of a specific region of bacterial 16S rRNA genes, which are highly conserved across bacterial species but contain variable regions that can be used for taxonomic classification. 14 The ideal approach to the study of the gut microbiota and the pathogenesis of neuroimmune diseases is to study untreated patients who are in the early stage of the disease. However, owing to diagnostic and therapeutic imperatives, many studies of patients with MS or NMOSD have involved those treated with steroids and/or disease-modifying therapies, and there has been a lack of research into the fecal microbiota of patients with ON alone, despite it being an early manifestation of various diseases.
In this study, we aimed to characterize the gut microbiota of patients with various types of ON (T-ON, NMOSD-ON, and ION) and others with MG, and to compare these with the microbiota of healthy controls, by performing 16S rRNA gene sequencing of DNA isolated from fecal samples. The inclusion of MG in the study allows us to identify potential differences in the gut microbiota associated with ON and other neuroimmune disorders affecting distinct parts of the ocular system. The findings may offer insight into the relationships of the gut microbiota with ON and MG, but especially ON, which is often considered to be a prodromal, early, or mild form of autoimmune disease of the CNS. The identification of unique microbiota profiles may enable the early differentiation of these conditions, thereby enhancing diagnostic accuracy and potentially improving clinical outcomes.
Materials and methods
Selection criteria for the participants
We performed a prospective study that was approved by the Ethics Committee of Beijing Tongren Hospital, Capital Medical University (approval number TRECKY2018-081) and conducted in accordance with the principles of the Declaration of Helsinki, 1975, as revised in 2013. All the participants provided their written informed consent, and the data collected were de-identified. We have reported the study according to the STROBE guidelines.
Initially, patients aged 18–30 years who presented with a first episode of ON and were admitted within 2 weeks of onset were consecutively recruited, to mitigate the influences of age and other factors on the gut microbiota. Patients with acute ON who displayed enhancement of the optic nerve signal (unilateral or bilateral) on gadolinium‐enhanced fat‐suppressed magnetic resonance imaging (MRI) scans were included. Patients with perineuritis and/or neuroretinitis, and those who exhibited symptoms or clinical signs or had MRI findings suggestive of brain and spinal cord involvement, were excluded.
The participants were categorized as having T-ON (essentially IDON, which can be considered to be a forme fruste of MS), NMOSD-ON, or ION, on the basis of the clinical features of the disease, laboratory testing, and at least 12 months of follow-up. The diagnostic criteria were as follows. (1) T-ON: patients with ON who experienced complete or near-complete visual recovery within 8 weeks of disease onset, who tested negative for AQP4-IgG and MOG-IgG, and for whom other diseases had been ruled out. (2) NMOSD-ON: patients with ON who tested positive for AQP4-IgG using a cell-based assay, after the exclusion of other connective tissue diseases and the presence of other antibodies besides AQP4-IgG. (3) ION: patients with ON who tested positive for antinuclear antibodies (titer ≥1:320.) ± extractable nuclear antigens, but did not meet the diagnostic criteria for any connective tissue disease at the time of sample collection. Patients with ocular MG (OMG) who tested positive for acetylcholine receptor antibody were also recruited, but those with MG that involved muscles other than extraocular muscles were excluded. These patients had all received an initial diagnosis and had not been treated using any medication. Owing to the fluctuating clinical course that characterizes MG, which often has a non-acute onset, no restrictions were placed on the timing of onset of the disease in patients with MG.
All the participants were from northern China and had similar dietary habits, with none reporting changes in diet or medication during the 7 days before stool sample collection. The exclusion criteria were as follows: (1) the presence of symptoms, signs, or imaging evidence of CNS involvement, other than in the optic nerve and extraocular muscles at onset; (2) the use of antibiotics or probiotics within the 4 weeks preceding the study; (3) the administration of steroids, immunosuppressants, or immunoglobulin, and/or plasma exchange within the 3 months preceding the study; (4) severe malnutrition or infection; (5) addiction to drugs, alcohol, or smoking; (6) a history of gastrointestinal conditions, including constipation, diarrhea, and/or other bowel diseases within the year preceding the study; and (7) the presence of diabetes, hypertension, or other multisystem diseases.
Sample collection and storage
Stool samples were collected using containers provided to both the patients and healthy controls. The samples were delivered in person or by a relative on the day of collection, involving a median transit time of 3 hours (range: 1–5 hours). Upon receipt, the stool samples were immediately frozen at −80°C until DNA extraction.
DNA extraction and 16s rRNA sequencing
Microbial genomic DNA was extracted from the stool samples using an E.Z.N.A.® soil DNA kit (Omega Bio-tek, Norcross, GA, USA), according to the manufacturer’s instructions. The extracted DNA samples were assessed on a 1% agarose gel, and the DNA concentration and purity were determined using a NanoDrop 2000 UV-vis spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The V3–V4 regions of the bacterial 16s rRNA genes were amplified using specific primers (forward primer: 5′-CCTAYGGGRBGCASCAG-3′ and reverse primer: 5′-GGACTACNNGGGTATCTAAT-3′), and polymerase chain reaction (PCR) was conducted in triplicate using an ABI GeneAmp® 9700 PCR thermocycler (Applied Biosystems, Waltham, MA, USA). The PCR products were then extracted from a 2% agarose gel and purified using an AxyPrep DNA gel extraction kit (Axygen Biosciences, Union City, CA, USA), according to the manufacturer’s instructions, then quantified using a Quantus™ fluorometer (Promega Corp., Madison, WI, USA). Purified amplicons were pooled at equimolar concentrations and subjected to paired-end sequencing on an Illumina MiSeq platform (Illumina, San Diego, CA, USA) according to standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
Bioinformatics and statistical analysis
The 16S rRNA data were analyzed using QIIME and R (www.r-project.org). Alpha-diversity was assessed using the abundance-based coverage estimator (ACE) and the Chao1, Shannon, and Simpson indices. 15 Beta-diversity was evaluated using Bray–Curtis distance matrices and visualized using principal coordinate analysis (PCoA). Taxonomic assignments were conducted using the GreenGenes database for 16S rRNA. Significant differences in bacterial richness were identified using linear discriminant analysis (LDA) effect size (LEfSe). Taxa with a log (LDA score) ≥4 and p < 0.05 were considered to be significantly enriched. Analysis of variance (ANOVA) was used to identify significant differences in the abundances of bacterial taxa among the groups.
SPSS version 27 (IBM Corp., Armonk, NY, USA) was used for the other statistical analyses. The Kolmogorov–Smirnov test was used to assess the normality of continuous datasets. Normally distributed continuous datasets are expressed as the mean and standard deviation (SD), non-normally distributed variables are expressed as median and interquartile range (IQR), and categorical variables are expressed as frequencies and percentages. Comparisons between multiple groups were made using the Kruskal–Wallis test, and comparisons of the distribution patterns of categorical datasets were made using the chi-square test. The level of statistical significance was set at p < 0.05.
Results
Study sample and stool sample collection
Stool samples were collected from 173 patients (137 with ON and 36 with MG) at the Neurology Department of Beijing Tongren Hospital, Capital Medical University, between 1 February 2018 and 30 December 2020. All the samples were obtained within 2 days of admission to the ward. After applying the specific inclusion and exclusion criteria, and a 12-month follow-up period, samples from 45 patients with ON, 13 patients with MG (Group M), and 20 age- and body mass index-matched controls (Group C) were analyzed. On the basis of the diagnostic criteria defined for this study, the patients in the ON group were placed into T-ON (Group T; n = 20), NMOSD-ON (Group N; n = 4), or ION (Group I; n = 11) groups. Detailed information regarding the participants is provided in Figure 1 and Table 1.
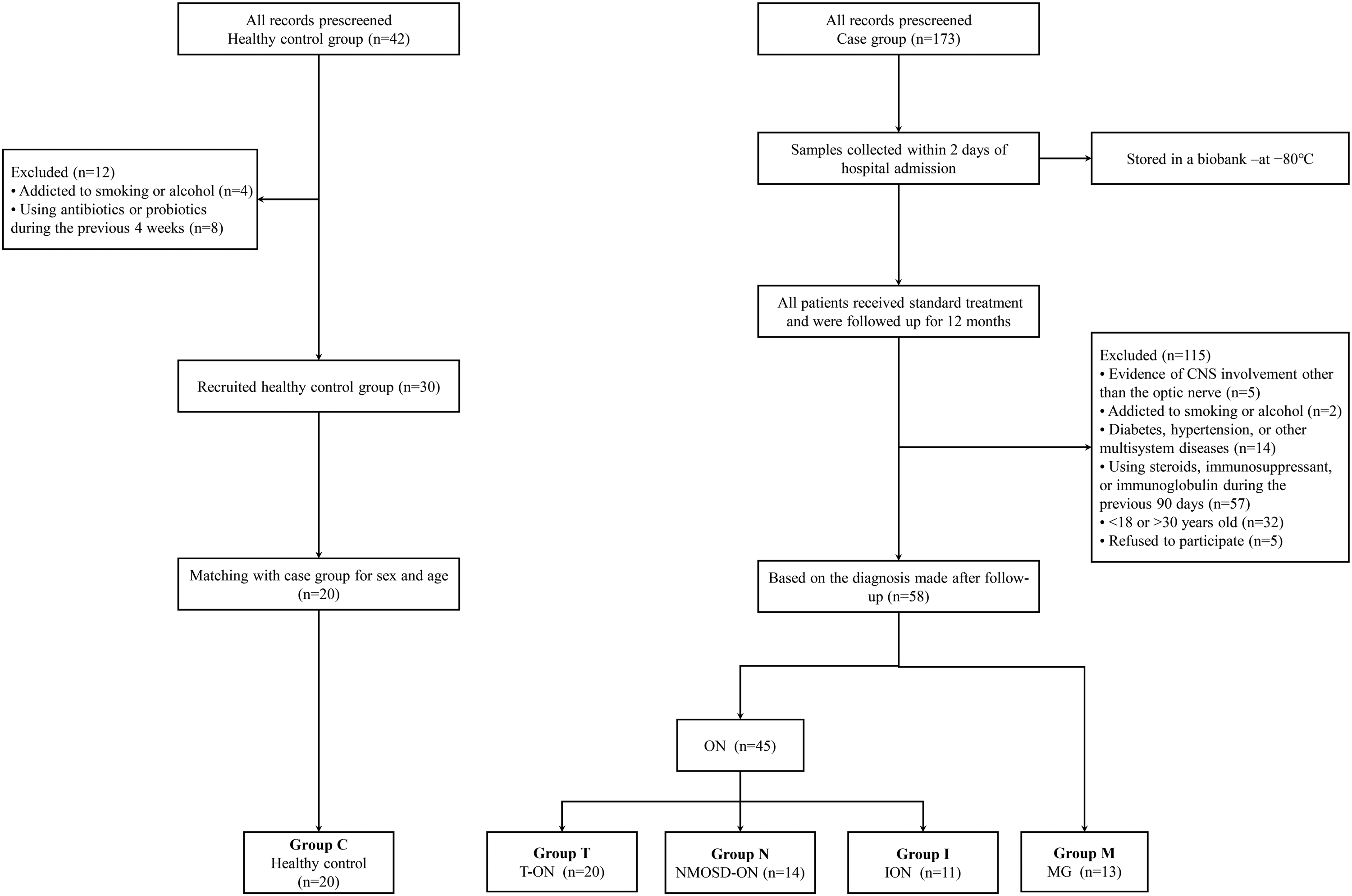
Flowchart of participant selection and categorization. ION, inflammatory optic neuropathy; MG, myasthenia gravis; NMOSD, neuromyelitis optica spectrum disorder-associated optic neuritis; ON, optic neuritis; T-ON, typical optic neuritis.
Comparison of the demographic and clinical characteristics of the patients with MS-ON, NMOSD-ON, ION, and MG and healthy participants.
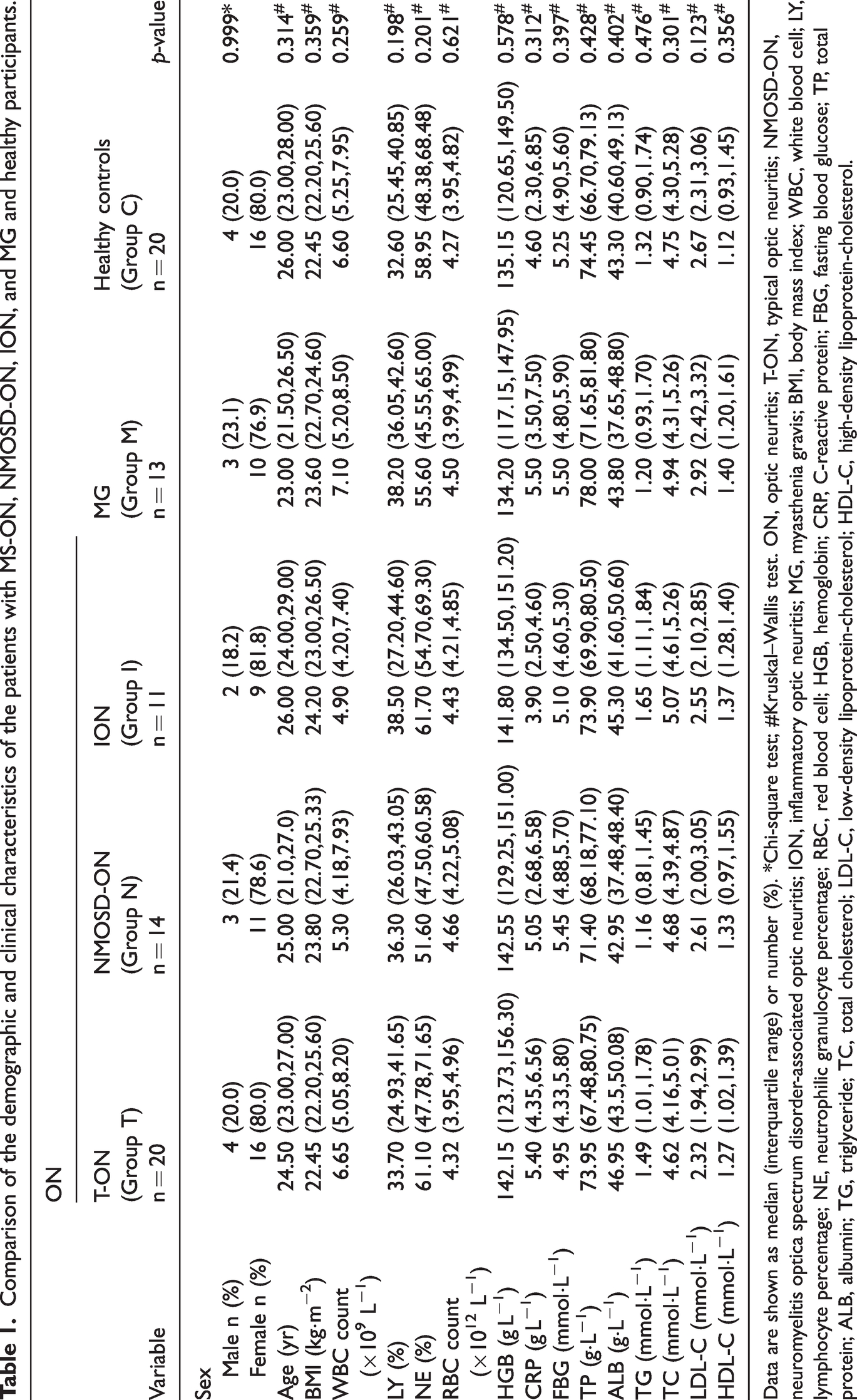
Data are shown as median (interquartile range) or number (%). *Chi-square test; #Kruskal–Wallis test. ON, optic neuritis; T-ON, typical optic neuritis; NMOSD-ON, neuromyelitis optica spectrum disorder-associated optic neuritis; ION, inflammatory optic neuritis; MG, myasthenia gravis; BMI, body mass index; WBC, white blood cell; LY, lymphocyte percentage; NE, neutrophilic granulocyte percentage; RBC, red blood cell; HGB, hemoglobin; CRP, C-reactive protein; FBG, fasting blood glucose; TP, total protein; ALB, albumin; TG, triglyceride; TC, total cholesterol; LDL-C, low-density lipoprotein-cholesterol; HDL-C, high-density lipoprotein-cholesterol.
Alpha- and beta-diversities of the gut microbiota of the patients and healthy controls
In microbiome studies, operational taxonomic units (OTUs) are used to describe clusters of sequences grouped by similarity, which serve as proxies for microbial species. Reads refer to individual DNA sequences obtained from high-throughput sequencing, which are then analyzed to identify and classify the OTUs present within a sample. 16 To characterize the composition of the microbial communities, we analyzed the OTUs and reads across the various disease groups. The OTUs and reads exhibited similarities between samples representing the various disease groups, but the mean abundance of OTUs was higher in the patients than in the healthy individuals (Figure 2(a)). Principal components analysis (PCA) based on OTU abundance revealed significant overlaps in the compositions of the bacterial communities among the groups, with principal component (PC)1 and PC2 explaining 18.93% and 12.78% of the variance, respectively (Figure 2(b)).The ACE and Chao1 indices, reflecting the total numbers of OTUs, were also significantly higher in the patients than in the healthy controls (Figure 2(c)–(d)), whereas the Simpson and Shannon indices, representing species richness and evenness, showed no statistically significant differences between the patients and controls (Figure 2(e)–(f)). In addition, no significant differences in alpha-diversity were found among Groups M, T, N, and I. To further characterize the differences in the species diversity among the groups, we used the beta-diversity distance matrix. A heatmap generated according to pairwise comparisons revealed significant disparities in species composition between the control and disease groups (Figure 2(g)). Moreover, a PCoA plot demonstrated separate clustering of samples from the healthy controls and patients (Figure 2(h)). These data suggested that the microbial communities were similar for the various disease subgroups, but differed from those of healthy controls.

Microbial diversity in patients with optic neuritis (ON) and related disorders. (a) Bar chart showing the operational taxonomic units (OTUs) present in all the samples, with an overlaid line graph showing the number of reads. (b) Principal components analysis (PCA) plot, illustrating the beta-diversity of the samples from the various groups. Each point represents a sample and colors indicate the various groups. (c, d) Abundance-based coverage estimate (ACE) and Chao1 indices, respectively, reflecting the species richness of the groups. (e, f) Simpson and Shannon indices, respectively, reflecting the species richness and the evenness of the groups. (g) Heatmap, generated to visualize the pairwise discrepancies in species composition among the groups. Darker shades correspond to greater discrepancies and (h) results of principal coordinates analysis (PCoA), reflecting the beta-diversity of samples from the control and disease groups. Each point represents a sample, and they are color-coded according to the group. PCO1 (13.81% variance) and PCO2 (11.68% variance) were the principal coordinates of the sample distribution, reflecting differences in the composition of the microbial communities between the samples. C, control; N, NMOSD-ON (patients with optic neuritis and neuroinflammatory disease); I, ION (inflammatory optic neuritis); T, T-ON (typical optic neuritis); M, MG (myasthenia gravis).
Composition of the gut microbiota of patients and healthy controls
To further investigate the microbial compositions of the groups, we analyzed the distributions of microbial taxa at the phylum and genus levels. At the phylum level, Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria predominated, but there was significant variability. For example, the proportion of Actinobacteria varied from 5.4% to 12.3% across the control group and disease subtypes (Figure 3(a)). At the genus level, we focused on 17 bacterial genera. The dominant bacterial genera differed among the groups: in the healthy controls, Bacteroides, Faecalibacterium, Bifidobacterium, Blautia, and Roseburia were most prevalent, whereas in the patient groups, the proportions of these differed, indicating potential disease-associated dysbiosis. Each patient group exhibited a unique microbiome, with variations in the prevalences of key genera, such as Bacteroides, Faecalibacterium, and Ruminococcus, indicating that there is a complex interaction between host health status and gut microbial composition (Figure 3(b)).

Compositions of the gut microbiota of healthy controls and patients. (a) Distribution of microbial phyla across the healthy control and patient groups. Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria predominated. (b) Relative abundances of 17 critical bacterial genera in the gut microbiota of the healthy control and patient groups. In the healthy control group, the dominant genera included Bacteroides (28.4%), Faecalibacterium (13.3%), Bifidobacterium (12.5%), Blautia (7.6%), and Roseburia (6.2%). The patient groups exhibited distinct microbial compositions: the I group was primarily colonized by Bacteroides (16.1%), Faecalibacterium (14.9%), Prevotella (11.0%), Roseburia (6.4%), and Ruminococcus (6.2%); the N group was primarily colonized by Bacteroides (17.3%), Faecalibacterium (15.2%), Ruminococcus (10.5%), Blautia (10.0%), and Bifidobacterium (6.1%); the T group was primarily colonized by Bacteroides (21.2%), Faecalibacterium (17.1%), Roseburia (7.4%), Ruminococcus (6.9%), and Blautia (6.4%); and the M group was primarily colonized by Faecalibacterium (13.6%), Ruminococcus (12.2%), Prevotella (10.4%), Bacteroides (7.6%), and Roseburia (6.5%). C, control; N, NMOSD-ON (patients with optic neuritis and neuroinflammatory disease); I, ION (inflammatory optic neuritis); T, T-ON (typical optic neuritis); M, MG (myasthenia gravis).
Abundances of specific microbial taxa in healthy controls and patients
To identify the microbial taxa responsible for the overall differences between the groups, we used LEfSe, focusing on the genus-level differences. As shown in Figure 4(a), taxa such as Veillonellaceae and Megamonas were more abundant in the I group, whereas a variety of taxa, including Bifidobacterium and Bacteroides, were more abundant in the C group. The M group was characterized by a predominance of Ruminococcus and Megamonas vs. the C group (p < 0.01; Figure 4(b)). In the N group, Lactobacillus and Lactobacillaceae were much more abundant than in the C group (p < 0.01; Figure 4(c)). Finally, there was a predominance of taxa such as Bacilli and Lactobacillales in the C group vs. the T group (p < 0.01; Figure 4(d)). However, no significant differences in the gut microbiota of the disease subgroups were identified. Further analysis of the abundances of bacterial families revealed that the Carnobacteriaceae family was significantly less abundant in all the participants with ON than in those with MG (p < 0.01; Figure 4(e)). However, separate comparisons between the M group and the other disease groups did not identify any significant differences in the abundances of bacterial taxa. In addition, there were no significant differences among the N, I, and T groups.
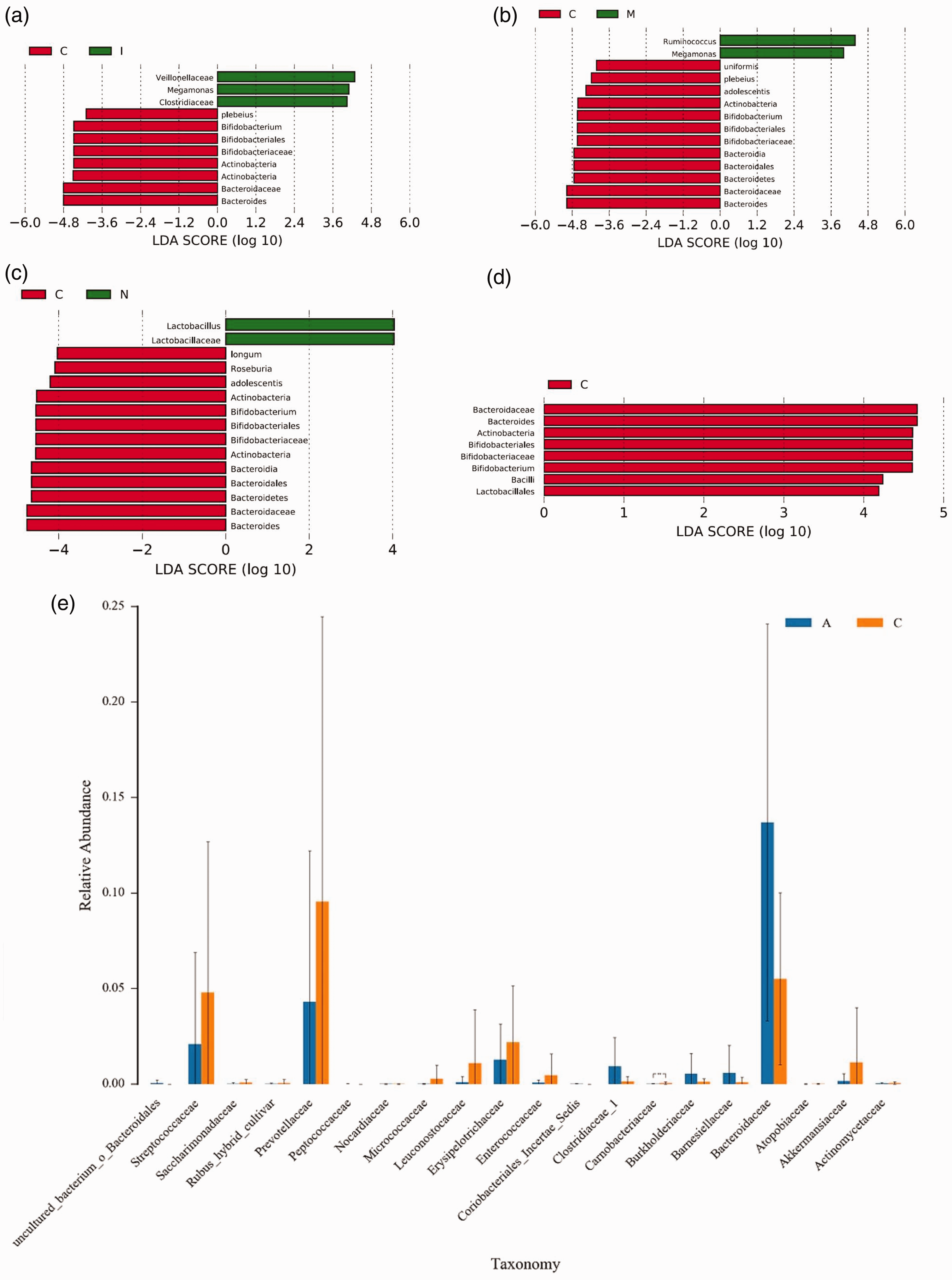
Results of the linear discriminant analysis effect size (LEfSe) and analysis of variance (ANOVA), revealing differences in microbial abundance. (a–d) Differential abundance of microbial taxa, revealed using LefSe. Comparisons between the C and I group (a), M group (b), N group (c), and T group (d), showing the taxa with significantly different abundances. Taxa that were more abundant in each patient group are indicated by green bars (positive LDA scores) and those that were more abundant in healthy controls are shown in red (negative LDA scores) and (e) relative abundances of microbial taxa at the family level in patients with MG (yellow) and ON (blue) patients. The Carnobacteriaceae family was more abundant in patients with MG than in those with ON in the N, I, and T groups. **p < 0.01. A, patients with ON; C, patients with MG; LDA, linear discriminant analysis; C, control; I, inflammatory optic neuropathy; M/MG, myasthenia gravis; NMOSD, neuromyelitis optica spectrum disorder-associated optic neuritis; ON, optic neuritis; T, typical optic neuritis.
Discussion
Although alterations in the gut microbiota have been recognized to be important factors in neuroimmune diseases, such as MS, NMOSD, and MG, there have been few studies of the gut microbiota in patients with ON or OMG alone, which could be considered to be mild or prodromal phases of these diseases.17,18 Although previous studies have included untreated patients, most have also involved patients who were being treated with steroids and/or immunosuppressants. Here, we present the first clinical study to include only untreated patients.
In the present study, by performing 16S rRNA sequencing of fecal samples obtained from patients with various neuroimmune diseases (T-ON, NMOSD-ON, ION, and MG) and healthy individuals, we have identified a higher abundance of gut microbes and distinct community compositions in patients with these diseases, compared to healthy controls. However, comparisons among patients with NMOSD-ON, ION, T-ON, or MG using PCA and PCoA revealed that their gut microbiota compositions were similar. Notably, distinct microbial taxa were overrepresented in the disease groups vs. the C group, including greater abundance of Veillonellaceae and Megamonas in the I group, of Ruminococcus and Megamonas in the M group, and of Bacilli and Lactobacillales in the C group vs. the T group. These results suggest that specific gut microbes may be involved in the pathogenesis and progression of neuroimmune diseases and may represent potential biomarkers of each disease and therapeutic targets. These findings suggest that, although these neuroimmune diseases may have common effects on the gut microbiota, relative to healthy individuals, certain microbial taxa may show disease-specific patterns of abundance. Numerous studies have demonstrated the involvement of specific gut microbes in mechanisms such as protein mimicry19,20; however, a more plausible interpretation of the present findings is that immune system dysregulation may broadly influence the gut microbial composition of patients with these conditions. These data support the potential for the use of microbial markers as general indicators of disease, while also suggesting that some of the microbial changes may reflect the influence of specific diseases.
The gut microbiota in NMOSD-ON
In recent years, there have been a number of studies of the association between the gut microbiota and NMOSD. Cree et al. identified a close association between AQP4-IgG-associated NMOSD and abundant Clostridium perfringens in the gut. 19 They proposed that AQP4-specific T cells cross-react with the ATP-binding cassette transporter of C. perfringens, which has significant sequence homology with AQP4-IgG. In addition, an abundance of Clostridium bolteae was associated with AQP4-IgG-positive NMOSD in Indian patients, when compared with seronegative patients, but this was absent in healthy controls.20,21 These findings suggest that these organisms may play a causal role in the immunopathogenesis of NMOSD in susceptible individuals.
In the present study, similar to the study by Gong et al., we did not find Clostridium perfringens or Clostridium bolteae in Chinese patients with AQP4-IgG-positive NMOSD. 22 However, in contrast to this study, in which an abundance of Streptococcus was identified, we identified low expression of the genus Roseburia (Class Clostridia and Family Lachnospiraceae) in the NMOSD-ON group. The Roseburia genus comprises five well-characterized species (Roseburia intestinalis, Roseburia hominis, Roseburia inulinivorans, Roseburia faecis, and Roseburia cecicola), all of which produce the short-chain fatty acids acetate, propionate, and butyrate. Roseburia has been shown to mitigate intestinal inflammation and maintain energy homeostasis by producing metabolites, 23 and previous studies have shown a significantly lower abundance of Roseburia intestinalis in patients with inflammatory bowel disease and demonstrated its anti-inflammatory effects in dextran sulfate sodium-induced colitis. 24 The lower expression of the genus Roseburia in patients with NMOSD-ON may contribute to a reduction in the anti-inflammatory effects of their metabolites.
The gut microbiota in ION
The overrepresented gut microbial taxa in patients with ION exclusively belonged to the order Clostridiales (class Clostridia), which has been implicated in various psychiatric disorders. A depletion of Clostridiales may lead to dysfunction of intestinal amino acid metabolism and lower production of short-chain fatty acids. 25 Gargari et al. have reported that the fecal abundance of Clostridiales reflects the symptoms of patients with irritable bowel syndrome, with greater abundance of Clostridiales in patients with the diarrhea type, which is characterized by enhanced colonic fermentation, than in those with the constipation type of IBS. 26
Many chronic inflammatory diseases result from dysregulated immune responses to autoantigens or environmental antigens. The gut microbiota has been established to be a key regulator of the pathogenesis of various immune-related conditions. For instance, the microbial composition of patients with early rheumatoid arthritis has been shown to involve a reduction in the abundances of the families Bifidobacterium and Bacteroides, along with greater abundance of the genus Prevotella.27,28 In patients with systemic lupus erythematosus, a prototypical systemic autoimmune disease, Hevia et al. showed a significantly lower Firmicutes/Bacteroidetes ratio compared to healthy individuals. 29 In the I group in the present study, we identified lower abundance of the genera Bifidobacterium (family Bifidobacteriaceae) and Bacteroides (family Bacteroidaceae) compared with normal controls, consistent with the findings of previous studies. Both Bifidobacterium and Bacteroides are candidate probiotics, because of their potential beneficial effects on host health. Notably, we did not identify greater abundances of other bacterial taxa that promote inflammation.
The gut microbiota in T-ON
In contrast to white patients with MS, who typically demonstrated greater abundance of Akkermansiaceae and Methanobacteriaceae and lower abundance of the Butyricimonas, Bacteroidetes, and Clostridia clusters,30,31 similar results have not been obtained in previous studies involving Chinese patients with MS.32,33 In the T group in the present study, essentially consisting of patients with IDON, we only identified lower abundance of the genera Bifidobacterium (family Bifidobacteriaceae) and Bacteroides (family Bacteroidaceae) than in the normal controls. Furthermore, the characteristics of the intestinal microbiota of both the Chinese and Caucasian patients studied previously30–34 were not replicated in the present study.
The gut microbiota in MG
Previous studies have identified increases in the abundances of taxa such as Bacteroidetes, Desulfovibrionaceae, and Pastereullaceae in patients with MG, along with decreases in the abundances of families such as Bifdobacteriaceae and Verrucomicrobiaceae and the phylum Firmicutes vs. healthy controls.11,35 However, in the present study, the characteristics of the gut microbiota of patients with MG were similar to those of patients with ION. Specifically, the abundances of the genera Bifidobacterium (family Bifidobacteriaceae) and Bacteroides (family Bacteroidaceae) were both lower than those of normal controls. In addition, the identification of overrepresentation of taxa in the order Clostridiales (class Clostridia) differs from the results of previous studies, which showed lower abundance of bacterial taxa belonging to the Lachnospiraceae and Ruminococcaceae families of the Clostridiales in patients with MG. 36
As described in the Results, the primary characteristic of the gut microbiota of patients with NMOSD-ON, T-ON, ION, and MG was lower abundance of probiotics such as the genera Bifidobacterium (family Bifidobacteriaceae) and Bacteroides (family Bacteroidaceae). There may be several possible explanations for this finding. Firstly, unlike in previous studies, the patients with ON in the present study only had optic nerve involvement. Although the etiology of ON may be similar to NMOSD, MS, or connective tissue diseases involving the CNS, the patients with ON had symptoms that were exclusively optic nerve-related or were prodromal and generally presented with milder symptoms. It is reasonable to speculate that in the early stages of these diseases, it is not increases in the abundances of some pathogenic bacteria, but rather a reduction the abundance of normal probiotics that manifests. In addition, according to the specific immune features of the disease, disease-specific gut microbial changes may emerge. Secondly, in the present study, all of the patients were in the acute stage of disease, within 2 weeks of onset, and had not received any steroids or immunosuppressive therapy. It has been suggested that, regardless of the putative mechanism, the gut microbial profiles of treated and untreated patients with MS differ. 37 Therefore, investigators should carefully consider whether patients have been treated or not, and even consider whether specific therapies could affect the gut microbiota differently, in order to mitigate the heterogeneity and minimize the presence of confounding factors in their studies.
In neuroinflammatory diseases, the temporal sequence of changes in the gut microbiota changes and CNS inflammation has not been characterized. Despite uncertainties regarding causality, an intriguing question arises: which occurs first, the dysregulation of probiotics, the reduction in intestinal microbes that inhibit inflammation, or the proliferation of pathogenic bacteria? Although definitive answers are lacking, there have been notable findings in previous studies. Across various populations and ethnicities, there has been a consistent finding of significantly lower abundance of probiotic microbes in disease cohorts, whereas the reproducibility of findings regarding pathogenic bacteria has been lower. Furthermore, there are few targeted interventions targeting specific possible pathogenic bacterial taxa. Instead, diseases can be modified through supplementation. Notably, Rinaldi et al. have demonstrated the prevention of disease in a rat model of MG through the administration of Bifidobacterium and Lactobacillus probiotics, suggesting their therapeutic potential. 38 Differences in the abundances of Deltaproteobacteria and Faecalibacterium are also consistent with the involvement of the gut microbiota in the pathogenesis of MG, 39 and warrant further investigation with the aim of developing treatments.
The present study had several limitations. First, the sample size was relatively small, partly because the study was conducted at a tertiary referral center, where patients often receive various steroid treatments, which may have confounded the results. Second, the COVID-19 pandemic significantly affected the rate of accumulation of cases. Although ongoing clinical observations regarding patient prognosis and intestinal microbial characteristics at various stages of disease are underway, further patient recruitment will be necessary to increase the statistical power and generalizability of the findings. Furthermore, the recruitment of a larger cohort would allow for a more detailed analysis of the potential disease-specific microbial profiles.
In summary, the findings of the present study underscore the importance of investigating gut microbial alterations in neuroimmune diseases. We have shown a consistent decrease in the abundance of intestinal probiotics in various conditions, indicating a potential early imbalance in the gut microbiota. Further research is warranted to elucidate the precise mechanisms underlying these changes and explore their therapeutic implications.
Footnotes
Author contributions
TW and SC performed the studies, collected data, and drafted the manuscript. TW, SC, JY, HJ, and CL performed the statistical analysis and participated in the study design. TW, HJ, JP, XK, JW, and SC participated in the acquisition, analysis, or interpretation of the data and drafted the manuscript. All the authors read and approved the final version of the manuscript.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.