
Abstract
Extraskeletal orbital mesenchymal chondrosarcoma is an extremely rare and highly aggressive tumor. We herein report a case involving a woman in her early 20s diagnosed with orbital mesenchymal chondrosarcoma. This case report aims to increase recognition and understanding of this condition.
Background
Mesenchymal chondrosarcoma (MC) is a rare, high-grade sarcoma of bone and soft tissue first described by Lightenstein and Bernstein in 1959. 1 Only a small number of case reports involving orbital MC have been published, with the largest case series comprising fewer than 50 cases.1–3 Orbital MC accounts for only 1% to 10% of all chondrosarcomas.4,5 This tumor most commonly occurs in young adults during the second or third decades of life, with a slight predominance in women. It is more frequently found in the head and neck regions, including the orbit, brain, meninges, soft tissues of the face, and lower extremities. Based on its location, MC has been categorized into two distinct types: one occurring in skeletal muscle and soft tissue, and the other in the central nervous system and spinal cord. 6 We herein report a case of orbital MC in a young woman. This clinical entity should be considered as a differential diagnosis of orbital lesions.
Case report
A woman in her early 20s presented to the Department of Orbital Disease and Ocular Oncology at our hospital with a 2-month history of diplopia in the right eye. She had noticed a painless mass in the superonasal quadrant of the right orbit, which had progressively enlarged over the same period and limited upper right eye movement. The patient had no significant medical, personal, systemic, or family history.
On examination, her best-corrected visual acuity was 0.9 in both eyes. Intraocular pressure measured 16 mmHg in the right eye and 14 mmHg in the left eye using a non-contact tonometer. The red glass test indicated dysfunction of the right superior rectus muscle. Anterior segment and fundus examinations showed no significant abnormalities. A computed tomography (CT) scan of the orbit revealed a well-defined soft tissue density lesion approximately 25 × 14 × 23 mm in size, located in the superonasal quadrant of the right orbit. The lesion was found outside the right posterior bulbar muscle cone, with uneven density and calcification. Contrast-enhanced CT demonstrated nodular and patchy enhancement during the arterial phase, which decreased during the venous phase. Bone depression adjacent to the lesion was noted, along with compression and displacement of the right lateral rectus muscle and the right lacrimal gland (Figure 1).
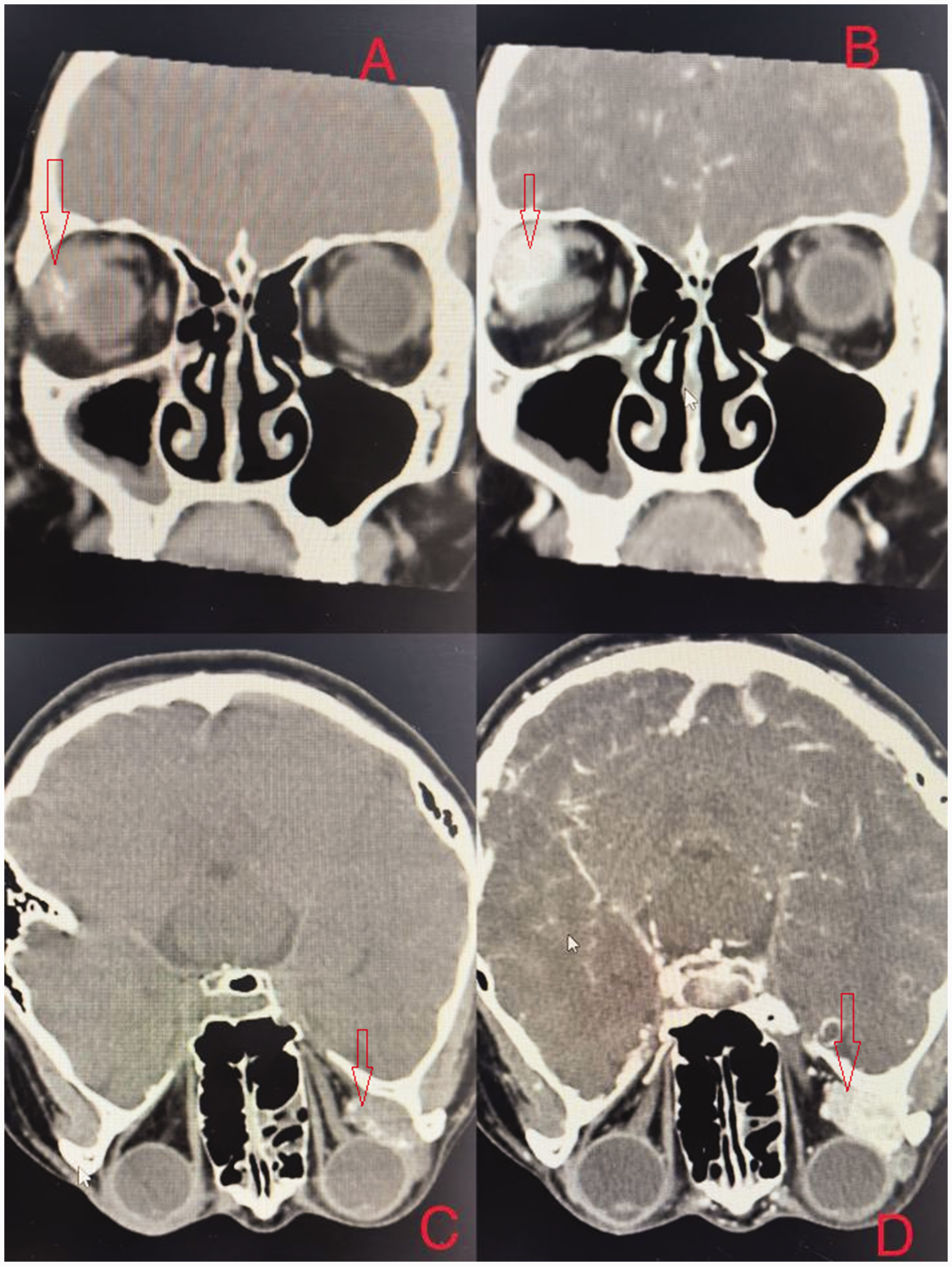
Imaging characteristics of the orbital lesion. (a) Computed tomography scans showed a well-defined soft tissue density lesion measuring approximately 25 × 14 × 23 mm in the superonasal quadrant of the right orbit. The density of the lesion was uneven, with localized calcification shadows visible. (b) Contrast-enhanced computed tomography showed nodular and patchy enhancement in the lesion. (c) The lesion was located outside the right posterior bulbar muscle cone and (d) the lateral rectus muscle and lacrimal gland were compressed and displaced.
Based on the imaging findings, cavernous hemangioma was initially considered as the diagnosis. Unfortunately, the patient declined magnetic resonance imaging (MRI) for personal reasons, and no MRI data were therefore available. After completing the necessary examinations and discussing the findings with the patient, informed consent for surgery was obtained. Two days later, the patient underwent orbital lesion resection under general anesthesia. Intraoperatively, the tumor was found to be attached to the lacrimal gland, necessitating excision of the tumor along with part of the lacrimal gland. The excised tissue was sent for histopathological examination. The intraoperative and postoperative courses were uneventful.
On gross examination, the mass was a grayish-brown nodule measuring 1.5 × 2.5 cm (Figure 2). Microscopically, the neoplasm consisted mainly of spindle cells with calcification and vascular lacunae (Figure 3). Immunohistochemical analysis revealed tumor cell positivity for S-100, transducin-like enhancer of split-1 (TLE1), B-cell lymphoma-2 (BCL2), and smooth muscle actin (SMA). The cells were negative for CK (AE1/AE3), epithelial membrane antigen (EMA), somatostatin receptor 2 (SSTR2), signal transducer and activator of transcription 6 (STAT6), and CD34. Molecular testing identified the HEY1-NCOA2 (H14;N13) gene fusion mutation. Based on the histology, immunohistochemistry, and molecular test results, the diagnosis of MC was confirmed.
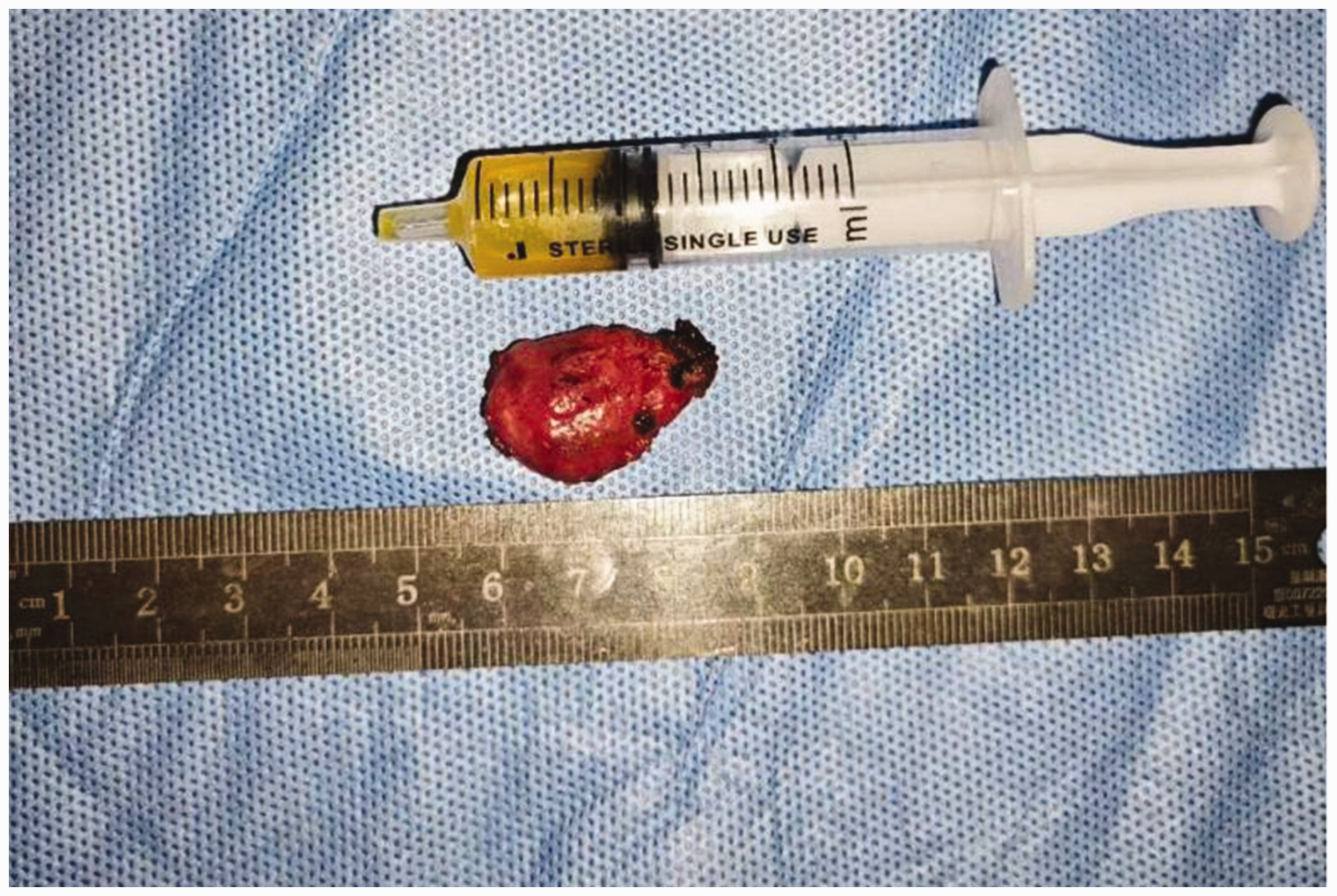
Gross appearance of the excised tumor. The tumor and part of the lacrimal gland were excised, measuring approximately 1.5 × 2.5 cm.

Histopathological findings of the tumor. Microscopically, the neoplasm consists mainly of spindle cells with calcification and vascular lacunae.
Discussion
MC originates from primitive cartilage-forming mesenchyme and accounts for 1% to 10% of all chondrosarcomas. 7 Extraskeletal MC of orbital origin is extremely rare, with most reports in the literature being case studies. The tumor typically occurs in young adults during the second or third decades of life, with a slight preponderance in women. The most common symptoms include progressive proptosis, decreased visual acuity, restricted motility, and ptosis.8,9 Orbital MCs are usually located in the intraconal space and characteristically appear as lobulated and well-defined soft tissue masses.8,10,11 These lesions may exhibit heavy calcification on CT imaging but more commonly reveal finely stippled, ring, or arc calcification, particularly in the central portion of the tumor. On contrast-enhanced CT, the lesions typically demonstrate moderate heterogeneous enhancement, with prominent delayed enhancement sometimes visible on dynamic contrast-enhanced CT. Localized bony erosion may also be observed in some cases. MRI characteristics of orbital MC include signal intensity lower than or equal to that of the brain on T1-weighted scans and isointensity to the brain on T2-weighted scans, with moderate enhancement after gadolinium administration. The calcified components of the tumor show low signal intensity on both T1- and T2-weighted MRI scans. Furthermore, contrast-enhanced MRI often reveals mild enhancement even within the calcified regions.8,12–14
Histologically, MC is relatively distinct. It demonstrates biphasic histology, characterized by sheets of undifferentiated, round or spindled mesenchymal cells interspersed with islands of hyaline cartilage. Typical immunohistochemical findings include positivity of the mesenchymal component for vimentin, Leu-7, and CD99 and positivity of the cartilaginous regions for S100 protein.4,5,7 Müller et al. demonstrated that the detection of type II collagen in the extracellular tumor matrix is a sensitive marker for identifying mesenchymal chondrosarcomas among small cell tumors both within and outside the bone. 15 In addition, Fanburg-Smith et al. reported Sox9 expression in 21 of 22 cases of MC (95.5%) 16 Both markers contribute to the diagnosis of MC.
Currently, surgery is the primary treatment for MC, with evidence suggesting improved survival in patients who undergo extensive surgical resection. 17 Postoperative adjuvant radiotherapy and chemotherapy can aid in achieving better local control.3,5,18 However, because of the small number of cases and limited data on prognosis, specific treatment options remain controversial. In this article, we have reported a rare case of orbital MC in a 24-year-old woman. This report adds to the limited available information on orbital MC and aims to provide further details about this condition to assist clinicians in recognizing this rare disease.
Footnotes
Acknowledgement
The authors are grateful to the Second Hospital of Jilin University for providing the platform for this case.
Author contributions
Conceptualization of the study: Qian LUO. Data acquisition: Qian LUO and Shuai WU. Manuscript preparation: Qian LUO. Manuscript revision: Qian LUO and Shuai WU. Both authors have read and approved the final manuscript.
Data availability statement
The data that support the findings of this study are available the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and publication of this article.
Ethics approval statements
Ethical approval to report this case was obtained from the Medical Ethics Committee of the Second Hospital of Jilin University (approval no. 311) on 17 July 2024. The case report has been de-identified by removing all personal details to ensure the patient’s privacy; thus, patient consent for publication was not required. The reporting of this study adheres to the CARE guidelines. 19
Funding
This research received no specific grant from funding agency in the public, commercial, or not-for-profit sectors.