
Abstract
Primary Sjögren’s syndrome (pSS) is an autoimmune disease that can cause gland dysfunction, leading to dry mouth and dry eyes as the main clinical manifestations. In addition, pSS may affect the gastrointestinal tract, skin, lungs, kidneys, nervous system, eyes, and other organs. We herein report a case involving a 38-year-old woman who presented with a 6-month history of dry mouth and eyes and a 10-day history of fever. She was diagnosed with pSS complicated by renal tubular acidosis, and her symptoms were alleviated after treatment with prednisone and mycophenolate. However, shortly after discontinuation of these medications outside the hospital, she experienced acute vision loss in both eyes. Bilateral uveitis secondary to pSS was diagnosed with the assistance of the Department of Ophthalmology, and the patient was treated with systemic steroids and topical ocular symptomatic therapy. After treatment, her systemic symptoms were relieved and her vision recovered. Although pSS complicated by renal tubular acidosis and uveitis is rare, it can lead to serious consequences such as abnormal renal function, visual impairment, and even blindness if not promptly treated. Steroid and immunosuppressive therapies are effective, and ophthalmology consultation should be performed if necessary for diagnosis and management.
Keywords
Introduction
Primary Sjögren’s syndrome (pSS) is a common autoimmune disease characterized by significant lymphocytic infiltration into the exocrine glands, leading to gland dysfunction due to the destruction of acinar and duct structures. The primary clinical manifestations of pSS are dry mouth and dry eyes. Its pathogenesis is associated with chronic inflammation of exocrine glands, influenced by genetic factors, immunodeficiencies, viral infections and environmental factors. In clinical practice, pSS not only damages the salivary and lacrimal glands but can also affect multiple systems and organs beyond the glands, including the gastrointestinal tract, skin, lungs, kidneys, nervous system, and eyes. Renal involvement typically presents as distal (type I) renal tubular acidosis (RTA), which may manifest as weakness or paralysis due to hypokalemia, nephrolithiasis, osteomalacia, systemic metabolic acidosis, slightly elevated serum creatinine, and mild proteinuria.1,2 When the eyes are involved, complications can include dry eye, uveitis, corneal melting and perforation, scleritis, retinal vasculitis, and optic neuritis. 3 Uveitis is a rare complication of Sjögren’s syndrome. In this report, we describe a case of acute bilateral uveitis in a patient with pSS complicated by RTA.
All patient details have been anonymized to ensure patient privacy, and written informed consent was obtained from the patient prior to treatment. Because all patient information has been de-identified, written informed consent for publication was not required. In addition, because of the nature of this study (case report), formal ethics committee approval was not required. The reporting of this study conforms to the CARE guidelines. 4
Case presentation
A 38-year-old woman presented to a local clinic with a 6-month history of mild dry mouth and eyes of unknown cause and a 10-day history of fever. She had no tooth loss, dental caries, parotid gland enlargement, dry eye, or eye congestion. Therefore, she did not seek medical attention. Ten days prior to her visit, she developed a fever of unknown cause with a peak temperature of 38.8°C, accompanied by chills, body aches, and fatigue. However, she reported no significant cough, expectoration, palpitations, shortness of breath, abdominal pain, diarrhea, abdominal distension, or urinary symptoms. After receiving 1 week of cephalosporin and penicillin infusions at the local clinic (specific drugs and doses unknown), the patient continued to experience intermittent fever, facial and lower extremity edema, upper abdominal pain, nausea, and vomiting. She was then admitted to our department for further investigation of the cause of her fever.
The patient had no history of allergies, alcohol or tobacco use, or drug abuse, and she had no relevant family history. Physical examination on admission revealed a body temperature of 38.7°C, heart rate of 105 beats/minute, respiratory rate of 20 breaths/minute, and blood pressure of 105/59 mmHg. She had palpable submental lymph node swelling, facial edema, a slightly dry tongue, and mild edema of the bilateral lower extremities. No swelling of the parotid gland, dental caries, tooth defects, or other abnormalities were noted.
Laboratory tests revealed the following (Table 1): positive anti-nuclear antibody (1:1000, nuclear particle type), positive anti-SSA antibody and anti-SSB antibodies (+++), anti-Ro52 antibody (+++), elevated IgG (17.8 g/L, reference range: 7.51–15.6 g/L), low hemoglobin (103 g/L, reference range: 115–150 g/L), elevated erythrocyte sedimentation rate (116 mm/hour, reference range: <38 mm/hour), and elevated C-reactive protein (93.128 mg/L, reference range: 0.068–8.200 mg/L). Electrolyte analysis showed hypokalemia (potassium 2.9 mmol/L, reference range: 3.5–5.3 mmol/L), mild hyperchloremia (chloride 107 mmol/L, reference range: 99–110 mmol/L), elevated creatinine (155 mmol/L, reference range: 30–90 mmol/L), and low carbon dioxide (18.9 mol/L, reference range: 21–31 mol/L). Blood gas analysis indicated metabolic acidosis with a pH of 7.19, actual bicarbonate of 9.2 mmol/L (reference range: 21.3–24.8 mmol/L), standard bicarbonate of 10.9 mmol/L (reference range: 21.3–24.8 mmol/L), and negative base excess in both blood (−17 mmol/L) and extracellular fluid (−19 mmol/L, reference range: −3 to +3 mmol/L). The lactic acid level was also elevated at 2.34 mmol/L.
Results of laboratory tests performed during the patient’s hospitalization.

ALC, absolute lymphocyte count; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; HLA-B27, human leukocyte antigen B27; /, not checked.
Based on the combination of symptoms, laboratory test results, and physical examination findings, we suspected pSS. After a discussion with the patient, we performed a labial gland biopsy. The pathological results showed chronic inflammatory changes consistent with Chisholm grade 3 (one focal lymph node infiltration) (Figure 1). According to the 2016 ACR/EULLAR classification criteria, the patient was diagnosed with pSS and RTA. After 5 days of treatment with methylprednisolone (40 mg every 12 hours), potassium supplementation, and acid correction, the patient’s body temperature, creatinine, blood gas, and electrolyte levels returned to normal (Table 1). The methylprednisolone therapy was then switched to oral prednisone (40 mg once daily), and oral mycophenolate mofetil (0.5 g every 12 hours for 2 days) was added for anti-inflammatory immunotherapy. The patient was then discharged.

Chronic inflammatory changes in labial gland biopsy. Imaging examination showed chronic inflammatory changes consistent with Chisholm grade 3 (one focal lymph node infiltration).
It should be noted that the patient took the prednisone and mycophenolate mofetil for only 1 day after discharge. Three days after discharge from the hospital, she developed pain in both eyes, photophobia, and decreased visual acuity. An ophthalmologic examination revealed visual acuity of 1.2/20 in the right eye and 2/20 in the left eye, with normal intraocular pressure. No redness or swelling of the eyelids was observed, but conjunctival hyperemia, transparent corneas, and dust-like keratic precipitates (i.e., inflammatory cells or pigment deposited on the posterior surface of the cornea) were noted. The anterior chamber depth was normal, with aqueous flare and positive Tyndall effect; the iris texture remained clear. Exudation was seen in the pupil area, the pupils were round with sluggish light reflexes, the lenses were transparent, and vitreous opacity and retinal flattening were observed (Figure 2).
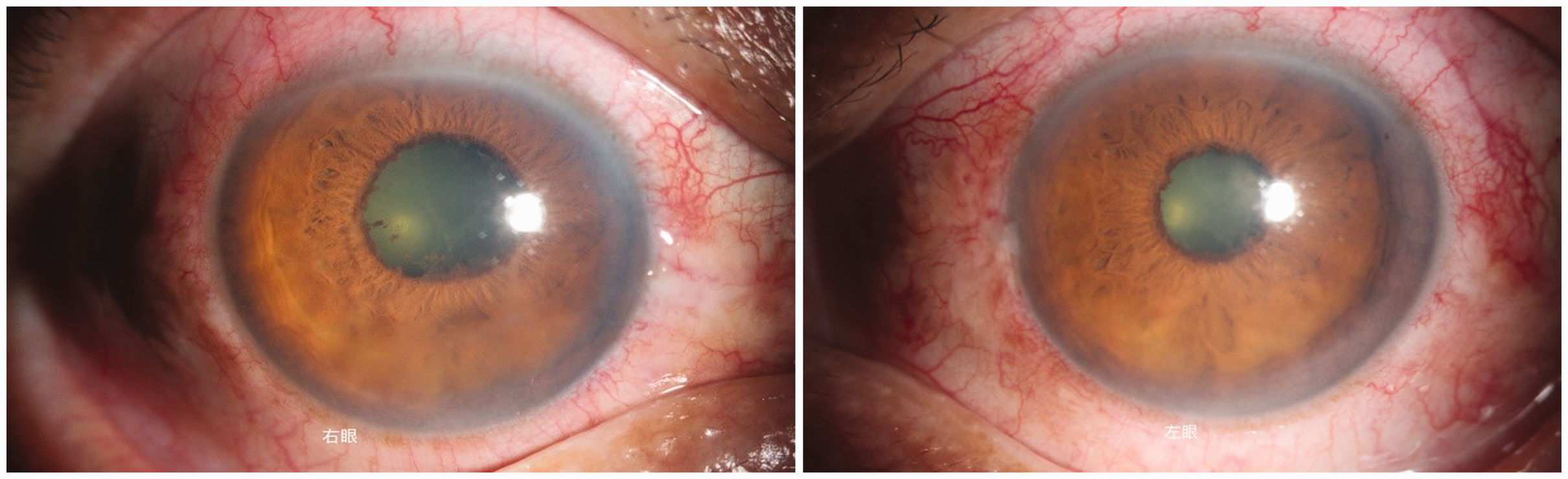
Anterior segment photograph of bilateral uveitis. Imaging examination revealed conjunctival hyperemia, dust-like keratic precipitates, aqueous flare, and exudation in the pupil area.
The final diagnosis was pSS complicated by RTA and acute bilateral uveitis. Treatment included oral prednisone (50 mg once daily), oral potassium citrate (1.45 g three times daily), tobramycin/dexamethasone eye drops, pranoprofen drops, and lacrimal duct flushing. After 8 days of treatment, the patient’s visual acuity recovered. The prednisone therapy was continued after discharge, and the dose was gradually reduced; however, the patient refused to resume immunosuppressive therapy because of concerns regarding side effects. Regular follow-up in the ophthalmology clinic showed no recurrence of symptoms within 2 months. At the last follow-up, the visual acuity was 20/20 in both eyes. The patient was subsequently lost to follow-up because she did not return to our hospital, and her contact number was incorrect.
Discussion
pSS is a chronic autoimmune disease that can affect various body systems. In addition to dry mouth and dry eyes due to impaired gland function, approximately one-third to one-half of patients develop systemic organ involvement, 5 affecting the joints, muscles, respiratory system, digestive system, kidneys, central nervous system, circulatory system, and other organ systems. 6 The reported prevalence of renal disease in patients with pSS is approximately 5%, with chronic tubulointerstitial nephritis being the predominant form of renal involvement. This condition typically presents as distal (type I) RTA, characterized by dysfunction of the cortical collecting ducts and impaired acid secretion by alpha-intercalated cells. Patients with pSS exhibit weakness or paralysis due to hypokalemia, nephrolithiasis, and osteomalacia, and they may also develop systemic metabolic acidosis, mildly elevated serum creatinine, and mild proteinuria.1,2 Our patient had metabolic acidosis, elevated creatinine, and hypokalemia, strongly suggesting tubulointerstitial nephritis despite the lack of pathological confirmation due to the patient’s refusal to undergo a renal biopsy.
Uveitis, when present, can be categorized based on the affected anatomical site: anterior uveitis (iris, ciliary body), intermediate uveitis (peripheral retina, vitreous), posterior uveitis (retina, retinal pigment epithelium, choroid, vitreous, optic nerve head) or panuveitis (all ocular tissues). Symptoms may include pain, photophobia, floaters, and decreased vision, and blindness may occur in severe cases. 7 The etiology of uveitis can be infectious or noninfectious, the latter of which is considered to be related to autoimmunity. 8 Autoimmune-related uveitis is relatively common in conditions such as seronegative spondyloarthropathy, Behçet’s disease, and sarcoidosis, among others. 9 Uveitis associated with Sjögren’s syndrome is a relatively rare but serious complication. There are no corresponding statistical data on its incidence. Its pathogenesis remains unclear but is currently believed to be related to autoimmunity.10–12 Previous studies have shown that circulating immune complexes are associated with both Sjögren’s syndrome and acute anterior uveitis.13,14 The pathogenesis of uveitis in pSS is believed to involve immune-mediated progressive and irreversible damage to photoreceptors caused by the involvement of regulatory B cells and autoreactive T cells, eventually leading to visual impairment and even blindness.9,15–17
Regarding previously published reports of Sjögren’s syndrome complicated by RTA and uveitis, we identified only one relevant case. Sugitani et al. 18 reported a case of tubulointerstitial nephritis and uveitis caused by Sjögren’s syndrome, confirmed by renal biopsy. In our case, the patient had a well-established diagnosis of pSS and subsequently developed RTA and secondary uveitis. Although we strongly suspected tubulointerstitial nephritis, we were unable to confirm in pathologically because the patient refused a kidney biopsy.
In addition, Sugitani et al. 18 used only hormone therapy, whereas we implemented a treatment regimen combining hormones, immunosuppressants, and local ophthalmic treatment. This approach raises the question of whether similar patients should receive both immunosuppressants and local ophthalmic treatment during follow-up, a topic that still needs further research.
Tubulointerstitial nephritis and acute uveitis in patients with pSS may lead to a multiple serious adverse outcomes, and there are currently no specific treatment recommendations for these complications. Additionally, there is no standardized treatment for pSS-associated interstitial nephritis aimed at reducing interstitial inflammatory infiltration and slowing the production of autoantibodies characteristic of Sjögren’s syndrome-related tubulointerstitial nephritis. At present, slowing the progression of the disease remains challenging. 19
Corticosteroids are the most commonly used treatment, but various other immunosuppressants with or without corticosteroids have been employed. These include hydroxychloroquine, rituximab, cyclophosphamide, methotrexate, mycophenolate mofetil, and azathioprine.20–23 Acidosis and hypokalemia are the most common manifestations of interstitial nephritis,24,25 and most patients are treated with potassium and bicarbonate supplementation in conjunction with immunosuppressive drugs. However, it has also been shown that some patients may remain dependent on oral potassium and bicarbonate supplements even after receiving immunomodulatory therapy. This suggests that while immunosuppressive therapy is crucial in maintaining or improving renal function and reducing interstitial infiltration, it may not be effective in fully resolving hypokalemia and acidosis.26–29
When immune diseases such as pSS are complicated by RTA and uveitis, an ophthalmologist is usually required to participate in the diagnosis and treatment alongside a nephrologist. The primary goal of treatment is to reduce ocular inflammation and achieve disease remission, thereby reducing or preventing serious consequences of ocular complications, such as blindness. 30
Depending on the severity of uveitis and its threat to vision, the general treatment recommendations are as follows.31–38 First, topical or systemic steroids are usually used to induce remission and control inflammation. In some cases, early administration of immunosuppressive agents, tumor necrosis factor inhibitors, interleukin-6 inhibitors, and CD20 monoclonal antibodies is also recommended for active treatment. Second, low-dose steroids are recommended for maintenance (<3 drops/day of topical steroids for anterior uveitis, <7.5 mg/day of prednisolone for non-anterior uveitis). Third, in cases of relapse, treatment with oral and/or topical corticosteroids, such as triamcinolone acetonide or dexamethasone administered periocularly or intravitreally, is advisable to maintain remission. Finally, discontinuation of therapy can be considered after achieving sustained long-term remission by gradually reducing all immunosuppressive treatments.
In our patient, pSS was initially complicated by RTA. After treatment with methylprednisolone (40 mg every 12 hours), potassium supplementation, and acid correction, the patient’s body temperature, creatinine, blood gas, and electrolyte levels returned to normal. However, after discharge from the hospital, the patient did not adhere to the prescribed regimen of steroids and mycophenolate mofetil. The subsequent development of acute bilateral uveitis was considered to be related to the recurrence of the disease. After consulting with the ophthalmology department, the patient’s vision recovered following local steroid therapy and systemic treatment. Unfortunately, the patient was not followed up in the rheumatology department, and her follow-up condition therefore could not be tracked. This highlights a significant limitation of the present study. It is very important to combine the rheumatology, nephrology, and ophthalmology departments for appropriate management.
In summary, pSS complicated by RTA and uveitis is rare, and delays in treatment can lead to serious adverse outcomes. It is very important to coordinate care among the rheumatology, nephrology, and ophthalmology departments to ensure timely diagnosis, treatment, and follow-up. Such multidisciplinary management can reduce the occurrence of chronic complications and even lower the risk of mortality and improve patients’ quality of life.
Footnotes
Author contributions
Jing Zhao, Haiyan He, and Yanlin Zhang contributed to the manuscript writing, editing, and data collection. Yuan Jia and Mei Tian conceived the idea for the article. All authors have read and approved the final manuscript.
Data availability statement
All data generated or analyzed during this study are included in this published article.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This study was funded by Foundation of Zunyi Science and Technology Department (Serial Number: ZUNSHIKEHE HZ296(2023)).
Informed consent and ethics statements
All patient details have been anonymized to ensure patient privacy, and written informed consent was obtained from the patient prior to treatment. Because all patient information has been de-identified, written informed consent for publication was not required. In addition, because of the nature of this study (case report), formal ethics committee approval was not required.