
Abstract
Objective
Apart from the role of the retinoblastoma gene, the genomic events associated with poor outcomes in patients with ophthalmic tumors are poorly understood.
Methods
We retrospectively analyzed 48 patients with six types of ophthalmic tumors. We searched for high-frequency mutated genes and susceptibility genes in these patients using combined exome and transcriptome analysis.
Results
We identified four clearly causative genes (TP53, PTCH1, SMO, BAP1). Susceptibility gene analysis identified hotspot genes, including RUNX1, APC, IDH2, and BRCA2, and high-frequency gene analysis identified several genes, including TP53, TTN, and MUC16. Transcriptome analysis identified 5868 differentially expressed genes, of which TOP2A and ZWINT were upregulated in all samples, while CFD, ELANE, HBA1, and HBB were downregulated. Kyoto Encyclopedia of Genes and Genomes enrichment analysis indicated that the phosphoinositide 3-kinase (PI3K)-Akt and Transcriptional misregulation in cancer signaling pathways may be involved in ophthalmic tumorigenesis.
Conclusions
TP53 is clearly involved in ophthalmic tumorigenesis, especially in basal cell carcinoma, and the PI3K-Akt signaling pathway may be an essential pathway involved in ophthalmic tumorigenesis. RUNX1, SMO, TOP2A, and ZWINT are also highly likely to be involved in ophthalmic tumorigenesis, but further functional experiments are needed to verify the mechanisms of these genes in regulating tumorigenesis.
Introduction
Tumors pose a considerable threat to human health and are among the most common and serious diseases. Tumors can occur in almost any part of the body, including most of the tissues in the eye, except the lens. 1 Research on ophthalmic tumors has greatly contributed to the understanding of tumor pathogenesis.2,3 The retinoblastoma gene (RB1), as the first identified oncogene and one of the most extensively studied tumor suppressor genes, is well known in this context4,5; however, compared with retinoblastoma, other ophthalmic tumors have received less-intensive study, 6 mainly because of their complex origin and classification. 7 The current study aimed to explore the specific mutational profiles of ophthalmic tumors to provide a theoretical foundation for subsequent targeted clinical treatment.
In this study, we used exon sequencing (ES) and transcriptome sequencing (RNA-Seq) to identify somatic point or small insertion/deletion (indel) variants, copy-number variations (CNVs), susceptibility genes, and driver genes underlying disease transformation. We collected cancer tissue, paracancerous tissue, and peripheral blood samples from 48 patients with six types of ophthalmic tumors: basal cell carcinoma (BCC), meibomian gland carcinoma (MGC), squamous cell carcinoma (SCC), choroidal melanoma (CM), sebaceous gland carcinoma (SGC), and intradermal nevus (IN). We then used ES and RNA-Seq to identify somatic point or indel variants, CNVs, and fusion genes as the basis for disease transformation. The primary objective was to identify ophthalmic tumor driver genes that may serve as biomarkers for drug therapy and early disease screening, while also promoting a better understanding of the genetic basis of ophthalmic tumors. We emphasize the importance of incorporating multiple sequencing platforms to comprehensively characterize the various mutation classes.
Methods
Patient samples and type of sequencing
In this retrospective study, we obtained cancer tissue samples, paracancerous tissue samples, and peripheral blood samples from patients who underwent ophthalmic tumor surgery at the Eye Center of the Second Affiliated Hospital of Zhejiang University between June 2020 and November 2021. The study included patients with BCC, MGC, SCC, CM, SGC, and IN. Notably, patients with CM had their left eyeball surgically removed; the eyeball was incised after surgery and normal choroidal tissue was collected from the inside of the eyeball to serve as a control. ES was performed in all patients, while RNA-Seq was performed in a subset of patients. The somatic cell status of variants was determined in paracancerous tissue and/or peripheral blood control samples. This study was conducted in accordance with the Helsinki Declaration of 1975 as revised in 2013 and approved by the Human Research Ethics Committee of the Second Affiliated Hospital of Zhejiang University (20220321). The reporting of this study conforms to the STROBE guidelines.8,9 All patients provided written informed consent and received genetic counseling. All data from this study were analyzed anonymously.
Genetic analysis
Total DNA was extracted from patient samples using standard methods for ES. 10 We performed ES using the GenCap™ HUMAN WHOLE Exon Probe V4.0 (MyGenostics, Beijing, China), and exome libraries were sequenced on the GenCap™ Platform (MyGenostics). Sequencing metrics indicated that all samples had sufficient sequencing coverage to confidently call variants, with a mean coverage of at least 46× at each targeted base and more than 90% of targeted bases with at least eight independent reads. We used published algorithms to identify somatic variants. We also used an in-house method to detect CNVs in normal and tumor samples and assessed structural variants and genomic fusions using Manta. 11
RNA-Seq libraries were constructed with 1 μg of total RNA using an Illumina TruSeq total RNA kit in conjunction with the Ribo-Zero Gold rRNA depletion kit (Illumina Inc., San Diego, CA, USA). Quality control was carried out using an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The protocol was capable of identifying sense and antisense spliced fusion transcripts, as well as genomic breakpoints from intron-retaining precursor messenger RNA, which are usually absent in standard poly(A)-enriched RNA-Seq. Gene fusions and other genomic rearrangements were analyzed using in-house tools based on output provided by the STAR RNA-Seq mapper, and single-nucleotide variants (SNVs) and indels in the RNA-Seq data were detected using FreeBayes. 12 Differential gene expression analysis is described in the Supplemental Methods. Sequencing was performed on Illumina HiSeq2500 and NextSeq 500 instruments (Illumina Inc.).
Filtering strategy for potentially pathogenic variants
Annotation of variants is described in the Supplemental Methods. Indels in protein-coding regions and nonsynonymous and essential splice-site SNVs predicted to be deleterious using in silico functional prediction models, as well as fusions and CNVs, were retained and classified according to clinical relevance (Supplemental Methods). Missense-coding variants designated as deleterious were predicted to be damaging by at least three of the four functional prediction algorithms, indicating that they were likely to modify protein function. These variants also met minimum evolutionary conservation scores.
Classification systems for clinical relevance were adapted to enrich for potentially pathogenic variants. Retained deleterious variants were classified as: 1) likely pathogenic, 2) pathogenic, or 3) unknown variants. Briefly, clinically relevant variants were associated with ophthalmic tumors and occurred in genes listed in the Catalogue of Somatic Mutations in Cancer Gene Census. 13 These genes have high-level evidence as drivers of ophthalmic tumors and were hereafter termed cancer genes. Possibly relevant variants occurred in genes recurrently mutated in our cohort. Novel somatic fusions and recurrent CNVs were also considered possibly relevant.
Bioinformatics analysis
In this study, the pathological mechanisms of ophthalmic tumors were examined using various bioinformatics analyses. We classified the differentially expressed genes identified from the ES data and RNA-Seq data overlaid onto pathways defined by databases, namely the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology Consortium (GO), using the R package cluster Profiler.
Statistical analysis
Groups were compared using the Mann–Whitney rank sum test. Frequencies were compared using Fisher’s exact test and the χ2 test. Statistical analyses were performed using SPSS 26 (IBM Corp., Armonk, NY, USA). P < 0.05 was considered statistically significant.
Results
Analysis of pathogenic SNV and CNV variants based on exome sequencing
We obtained cancer tissue samples, paracancerous tissue samples, and peripheral blood samples from 48 patients who underwent ophthalmic tumor surgery, including six types of ophthalmic tumors, namely 27 cases of BCC, 15 cases of MGC, 1 case of SCC, 1 case of CM, 3 cases of SGC, and 1 case of IN. ES was performed on DNA from all 48 patients, while RNA-Seq was performed on RNA from 15 of the 48 patients (Figure 1 and Tables S1 and S2). The mean age of the patients was 43 years (range: 20–69 years), with 30 women and 18 men. Of the 48 patients included in this study, 32 patients (66.67%) carried a total of 42 suspected pathogenic candidate loci, including TP53 (17/42), PTCH1 (16/42), RB1 (4/42), SMO (3/42), BRAF (1/42), and BAP1 (1/42). Among the 19 patients with pathogenic candidate loci, 18 (37.5%) had BCC and carried 26 pathogenic loci, while one patient (2.08%) with CM carried one pathogenic locus. The most frequently mutated genes associated with BCC were PTCH1 (15/26), TP53 (8/26), and SMO (3/26). Thirteen mutations were identified in patients with lid adenocarcinoma, including TP53 (8/13), RB1 (4/13), and PTCH1 (1/13), but none of these mutations were phenotypically related. Mutations in BAP1 (1358_1359delAA) were found in CM, IN, and SGC, and BRAF and TP53 were also associated with these mutations, but none were consistent with their phenotypes (Table S3 and Figure S1).

Patient ophthalmic tumor classification and type of sequencing analysis. (a) The mean patient age was 41 years, 37.5% of patients were men and 62.5% were women and (b) All patient samples were tested by ES and 15 patient samples were tested by ES and RNA-Seq. ES, exome sequencing; RNA-Seq, transcriptome sequencing; n, number; IN, intradermal nevus; SCC, squamous cell carcinoma; CM, choroidal melanoma; SGC, sebaceous gland carcinoma; MGC, meibomian gland carcinoma; BCC, basal cell carcinoma.
According to The American College of Medical Genetics and Genomics guidelines, 165 candidate CNV variants were screened in 35 samples in this assay, including 145 in the main sample and 20 in the control, with no duplicate CNVs in the main and control samples. Fifty-five CNVs were associated with BCC, 72 with MGC, 32 with SGC, and six with SCC. Patients 2, 12, 31, 33, and 59 with BCC had clearly associated CNV fragments (Table S4).
ES analysis for screening gene-based tumor susceptibility genes
We identified 43 susceptibility genes in 44 samples, with hotspot genes including RUNX1 occurring six times, APC five times, IDH2 five times, and BRCA2 four times. Specifically, loci chr21:36164881 and chr21:36164870 of the RUNX1 gene occurred three times each, while locus chr5:112175007 of the APC gene occurred twice. Loci chr15:90634814 and chr15:90630685 of the IDH2 gene and locus 112175007 also occurred twice each. Mutations were also detected at loci chr15:90634814 and chr15:90630685 of the IDH2 gene twice each (Table S5).
High-frequency mutation loci and CNV analysis
High-frequency mutated genes have a higher mutation frequency than the background mutation frequency. In this study, TP53 had the highest mutation rate among all samples at 44%, followed by TTN 34%, and MUC16 31% (Figure S2). CNV variants were detected in each sample using CNV kit software, followed by the use of GISTIC 2.0 software to evaluate significant regions and to report the location of chromosomal coordinates, q-values values of the region, and the genes contained in the region. The distribution of high frequency CNV regions on chromosomes is shown in Figure S3, and the Oncoplot of high frequency CNV is shown in Figure S4. The significantly amplified and missing regions are presented in Table 1.
High frequency copy number variation amplifications and deletions.

Mutation profile of ophthalmic tumors based on transcriptome sequencing data analysis
In this transcriptome study, mRNA analysis was performed on a total of 19 paired samples from 15 patients (Figure 1 and Table S2). A total of 5868 differentially expressed genes were identified, including 2198 upregulated genes and 3670 downregulated genes (Figure S5). Further analysis of the top 30 upregulated and downregulated genes showed that TOP2A and ZWINT were consistently upregulated in all samples, while CFD, ELANE, HBA1, and HBB were all consistently downregulated (Figure 2).

Heat map of the top 60 differentially expressed genes.
KEGG pathway enrichment and GO functional enrichment analysis
The KEGG database is a comprehensive resource that integrates genomic chemistry and systemic functional information. Enrichment of KEGG pathways was conducted with a significance threshold of Padj < 0.05. Enrichment analysis of all differentially expressed genes initially revealed that the transcriptional misregulation in cancer and phosphoinositide 3-kinase (PI3K)-Akt signaling pathways may be involved in ophthalmic tumorigenesis, both of which were enriched in the upregulated group. TP53, which was widely identified in the pathogenicity analysis of ES, was involved in the PI3K-Akt signaling pathway (Figure 3). Comprehensive analysis of GO gene functions revealed that Biological Process focused on the positive regulation of cell adhesion, Cellular Component focused on the transcription regulator complex, and Molecular Function focused on transmembrane receptor protein kinase activity and transcription coregulator activity (Figure 4).

Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis. Vertical coordinates represent different pathways and horizontal coordinates represent proportion of genes mutated in the corresponding pathway compared with all genes in that pathway. Color indicates enrichment significance, with redder color indicating greater significance. Circles in the left vertical coordinate sorted according to GeneRatio value from largest to smallest, and right vertical coordinate sorted according to the p.adjust value from smallest to largest.
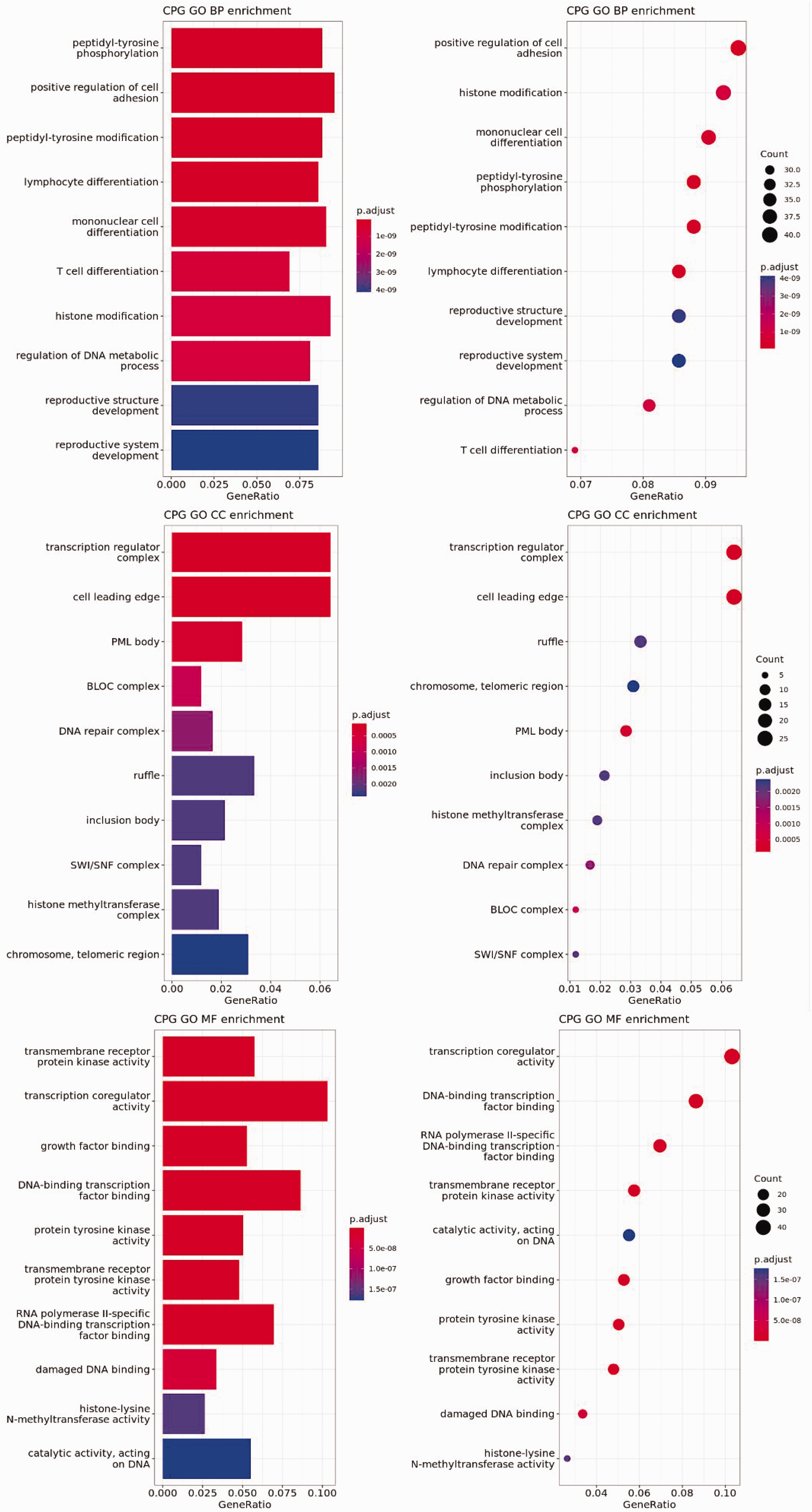
Gene Ontology functional enrichment analysis.
Driver gene screening and function prediction
Differential expression analysis was conducted to investigate the targeted pathogenicity genes, susceptibility genes, and high-frequency mutated genes in ES. Significant differences in the expression of BRCA2, PTCH1, TTN, TP53, RUNX1, MUC16, and SMO (P ≤ 0.05) were observed (Table S6). Notably, TP53, RUNX1, and SMO were highly associated with the ophthalmic tumor phenotype and may thus potentially serve as marker genes for ophthalmic tumors (Table 2).
Marker gene screening for ophthalmic tumors by exon-combined transcriptome sequencing analysis.

Discussion
In this study, we conducted a comprehensive analysis of six types of ophthalmic tumors (BCC, MGC, SCC, CM, SGC, IN) using ES and transcriptome approaches. The results suggest that these ophthalmic tumors have distinct molecular markers and combinations that may aid their accurate identification and discrimination by bioinformatics methods. We sequenced cancer tissues, paracancerous tissues, and peripheral blood, and observed that driver gene mutations were relatively common in cancer tissues at the DNA level, with some low-frequency mutations of non-driver genes in paracancerous tissues, and mutations were almost absent in peripheral blood, demonstrating a clear stepwise pattern. This pattern may be attributed to the planar distribution of skin cancers, where cancerous and paraneoplastic tissues are more easily distinguished for sampling, highlighting one of the characteristics of ophthalmic tumors.
Genetic tumor marker testing is crucial for the diagnosis and monitoring of tumors, including their early detection and screening, the differentiation of benign and malignant tumors, and the assessment of tumor efficacy and recurrence and metastasis monitoring. 14 The analysis of marker genes is thus paramount for the early diagnosis of ophthalmic tumors. In the present study, among the top 12 mutated genes from the whole ES data, 5 genes (TP53, PTCH1, SMO, BAP1, SUFU) were associated with BCC, 3 (TP53, PTCH1, RB1) with MGC, 2 (LEMD3, BRAF) with IN, and 3 (CDKN2A, SLX4, TGFBR2) with SCC, while two mutations in BAP1 were associated with CM, and SGC was only associated with TP53. Owing to their rarity, we only collected one sample each for IN, SCC, and CM; however, BRAF 15 mutation was previously shown to be associated with IN. CDKN2A 16 and TGFBR2 17 mutations were associated with SCC and BAP1 18 gene mutation was associated with CM. In addition, CNV analysis identified 165 pathogenic CNVs, with 5 CNVs clinically diagnosed as consistent with the diagnosis. Notably, large segments of CNVs were observed in several chromosomes, particularly in the regions of chromosomes 20, 9, 19, and 5, indicating high genomic instability and tumor malignancy in cancer cells. The findings suggest that pathogenic genes and CNVs can serve as molecular markers for ophthalmic tumors, enabling the early detection and differentiation of different cancer types. This information may improve the accuracy of bioinformatics methods for ophthalmic tumor diagnosis and monitoring, potentially contributing to improved patient outcomes.
We conducted marker gene analysis using susceptibility genes, high-frequency genes, and high-frequency CNVs. The analysis of susceptibility genes identified hotspot genes such as RUNX1, APC, IDH2, and BRCA2. Among these, medulloblastoma caused by the BRCA2 gene is one of the most common pediatric brain tumors, 19 with approximately 16% of pediatric brain tumors and 40% of pediatric cerebellar tumors being medulloblastomas. 20 The age of onset for medulloblastoma is bimodal, with peaks between 3 and 4 and between 8 and 9 years of age, respectively, with around 10% to 15% of medulloblastomas diagnosed in infants. In adults however, less than 1% of central nervous system tumors are medulloblastomas, with a peak between 20 and 34 years of age. Medulloblastomas are often associated with other syndromes, with 1% to 2% of patients having Gorlin syndrome (nevus-like basal cell carcinoma). 21 In the current study, sample 49 showed TP53 818G>A, Arg273His and samples 21, 22, and 50 showed RB1 1861C>A, Arg621Ser, and 1573G>A, Ala525Thr. Mutations in the TP53 tumor suppressor gene are found in around 50% of human cancers. 22 TP53 functions as a transcription factor that directly regulates the expression of approximately 500 genes, some of which are involved in cell cycle arrest/cell senescence, apoptotic cell death, or DNA damage repair (i.e., the cellular responses that together prevent tumorigenesis). 23 TP53’s related pathways include gastric cancer and RET signaling. RB1 is also a protein-coding gene, associated with diseases including retinoblastoma and small cell lung carcinoma.24,25
High-frequency gene analysis revealed several important genes, including TP53, TTN, and MUC16. TTN encodes myosin (Titin), which is expressed mainly in skeletal and cardiac muscle tissues and can cause a variety of cardiomyopathies when mutated. 26 MUC16 is a protein-coding gene associated with ovarian cysts and clear cell adenocarcinoma. High frequency CNV analysis identified multiple amplified and deleted regions, including 15q11.2, 1p36.21, 1q21.3, 7p11.2, and 5q31.3 amplifications, and 11q12.3, 6p21.32, 12p13.31, 17p13.1, and 8p11.23 deletions. Specifically, regions 12p13.31 and 8p11.23 were tentatively identified as pathogenic variants due to copy number deletions and the presence of haplo-dose-sensitive genes CHD4 and FGFR1, respectively. These variants have not been reported in the Decipher syndrome and OMIM syndrome databases. NSD3 was also reported in the dbvar case database for 8p11.23. 27 In addition, high-frequency CNV analysis identified several amplified regions, including 15q11.2, 1p36.21, and 1q21.3, and deleted regions, including 11q12.3, 6p21.32, 12p13.31, 17p13.1, and 8p11.23. Among these, the deletion of 12p13.31 (12: g.6344936_7194854del) was tentatively identified as a pathogenic variant due to its copy number deletion of approximately 849.918 Kb. This region contains the haplo-dose-sensitive gene CHD4, which has been reported in the dbvar case database for ATN1. Deletion of 8p11.23 (chr8: g.37909472_38965193del) was also tentatively classified as pathogenic due to its copy number deletion of approximately 1.06 Mb. This region contains the haplo-dose-sensitive genes FGFR1 and NSD3, which have been reported in the dbvar case database. In summary, the current research identified various genes and CNVs associated with specific diseases, such as TP53 with Li-Fraumeni syndrome and osteosarcoma, and TTN with cardiomyopathies. The study also highlighted potential pathogenic variants in the 12p13.31 and 8p11.23 regions because of their deletions of important haplo-dose-sensitive genes. These findings further our understanding of the underlying genetic causes of various diseases and may aid the development of more effective diagnostic and therapeutic approaches. 28
A total of 5868 differentially expressed genes were identified by transcriptome analysis, of which TOP2A and ZWINT were upregulated and CFD, ELANE, HBA1, and HBB were downregulated in all samples. TOP2A and ZWINT may be closely related to tumorigenesis. TOP2A encodes a topoisomerase that is closely related to DNA stability, and genomic instability caused by mutations in TOP2A is closely associated with tumorigenesis. 29 ZWINT encodes a protein found in tumor cell lines, which is closely related to the function of mitophagy. Musio et al. (2004) found that antisense oligonucleotide pairs INCENP (604411), ZWINT, and ZW10 inhibition led to the appearance of mitotic cells characterized by mitotic segregation, chromosome aneuploidy, and micronucleus formation. 30 Enrichment analysis of all differentially expressed genes first revealed that the Transcriptional misregulation in cancer and PI3K-Akt signaling pathways may be involved in ophthalmic tumorigenesis. They were all enriched in the upregulated group, and the TP53 gene, which was widely found in the pathogenicity analysis of ES, is involved in the PI3K-Akt signaling pathway.
There are some limitations to this study. This study involved only 48 patients, which is a relatively small sample size. Therefore, this sample size may not have been sufficient to capture the full spectrum of genetic variations associated with all types of ophthalmic tumors. Although the study identified several potential oncogenic genes and pathways, the specific functions and mechanisms of these genes and pathways require further experimental validation to confirm their roles in tumorigenesis.
Conclusions
In this study, TP53 was shown to be involved in ophthalmic tumorigenesis, particularly in BCCs, and the PI3K-Akt signaling pathway was identified as a critical pathway involved in ophthalmic tumorigenesis. RUNX1, SMO, TOP2A, and ZWINT are also likely to be involved in ophthalmic tumorigenesis, but further functional experiments are needed to confirm the mechanisms of these genes in regulating tumorigenesis.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605241258171 - Supplemental material for Integrative genomic analysis reveals cancer-associated mutations in patients with ophthalmic tumors
Supplemental material, sj-pdf-1-imr-10.1177_03000605241258171 for Integrative genomic analysis reveals cancer-associated mutations in patients with ophthalmic tumors by Fengjiao Zhu, Pengpeng Wang, Zhiyuan Zhang, Chunlei Yao, Yijie Wang, Juan Ye and Jian Wu in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605241258171 - Supplemental material for Integrative genomic analysis reveals cancer-associated mutations in patients with ophthalmic tumors
Supplemental material, sj-pdf-2-imr-10.1177_03000605241258171 for Integrative genomic analysis reveals cancer-associated mutations in patients with ophthalmic tumors by Fengjiao Zhu, Pengpeng Wang, Zhiyuan Zhang, Chunlei Yao, Yijie Wang, Juan Ye and Jian Wu in Journal of International Medical Research
Supplemental Material
sj-xlsx-3-imr-10.1177_03000605241258171 - Supplemental material for Integrative genomic analysis reveals cancer-associated mutations in patients with ophthalmic tumors
Supplemental material, sj-xlsx-3-imr-10.1177_03000605241258171 for Integrative genomic analysis reveals cancer-associated mutations in patients with ophthalmic tumors by Fengjiao Zhu, Pengpeng Wang, Zhiyuan Zhang, Chunlei Yao, Yijie Wang, Juan Ye and Jian Wu in Journal of International Medical Research
Footnotes
Acknowledgements
We wish to thank all the patients who participated in the study and all the staff at the clinic for their help in recruiting patients.
Author contributions
Jian Wu: Funding acquisition and Project administration; Fengjiao Zhu and Pengpeng Wang: Data curation, Formal Analysis, Software, Visualization, and Writing – original draft; Zhiyuan Zhang: Resources, Investigation. Chunlei Yao: Conceptualization, Supervision and Writing – review & editing; Jie Ii: Conceptualization, Project administration, Supervision and Writing – review & editing.
Availability of data and material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Declaration of conflicting interest
All authors declare no competing interests.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Jian Wu was supported by a grant from the National Key Research and Development Program of China [grant number 2019YFC0118403]. The funders were involved in the design of the study, the collection, analysis, and interpretation of the data, the writing of the manuscript, and the decision to submit the manuscript for publication.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.