
Abstract
Pulmonary blastoma (PB) is a rare, highly malignant tumor prone to distant metastasis and recurrence, and the prognosis of these patients is often poor. We report a case of metastatic PB with a good prognosis with the aim of providing data to support a clinical diagnosis and treatment. In December 2015, a 43-year-old male patient was admitted to our hospital because of a cough and blood-stained sputum. Positron emission-computed tomography showed massive high-density imaging in the lower lobe of the right lung, with a maximum cross-section of 76 × 58 mm. Thoracoscopic-assisted right lower lobectomy with lymph node dissection was performed. After 1 month, computed tomography showed a high possibility of metastasis. The patient then received docetaxel and cisplatin chemotherapy for a total of six courses. After chemotherapy, enhanced computed tomography showed considerable absorption of pleural effusion, and a left lobe pulmonary nodule was not detected. The postoperative pathological diagnosis was PB, and epithelial and mesenchymal differentiation components were observed. The patient continued to visit the hospital regularly for re-examination and imaging examinations. Currently, no signs of recurrence or distant metastasis have been detected.
Keywords
Introduction
Pulmonary blastoma (PB) is a rare and highly malignant tumor, with an incidence of less than 0.1% of all lung tumors. 1 PB is aggressive and may invade the mediastinum or have intrapulmonary metastasis. The prognosis of these patients is poor, with an overall 5-year survival rate of approximately 16%. 2 The clinical symptoms of PB are non-specific, including cough, hemoptysis, and chest pain, and approximately 40% of patients have no obvious symptoms in the early stage. The typical imaging findings are round or oval solid masses with smooth edges and lobed surroundings, but generally without “burr” formation. 3 When the tumor invades the bronchus, pleura, or pericardium, it may show corresponding imaging features, including obstructive pneumonia, pleural fluid, and atelectasis. 4 Therefore, PB is easily misdiagnosed or ignored clinically.
Pathology is the gold standard for the diagnosis of PB. The 5th World Health Organization classification of thoracic tumors proposes that PB, which is a subtype of sarcomatoid carcinoma, is a bidirectional differentiated tumor consisting of low-grade/well-differentiated fetal lung adenocarcinoma and primitive mesenchymal cells. 5 Specific mesenchymal differentiations (osteosarcoma, chondrosarcoma, or rhabdomyosarcoma) may also be present, but are not necessary for a pathological diagnosis. Cytokeratin (CK), carcinoembryonic antigen, epithelial membrane antigen, and thyroid transcription factor-1 (TTF-1) are diffusely strongly positive in epithelial components, β-catenin is positive in nuclear expression, and chromogranin A (CgA), synaptophysin (Syn), and vimentin can be focally expressed. Mesenchymal embryoid cells express vimentin and specific actin. 6
We report a case of PB with a good prognosis in which no recurrence or distant metastasis was detected 7 years after surgery and postoperative chemotherapy. Through the analysis of clinical data, pathological features, and genetic mutations, we attempted to examine the relevant factors affecting the prognosis of patients with PB to provide important data for supporting its clinical diagnosis and treatment.
Case presentation
Clinical data
In December 2015, a 43-year-old male patient was admitted to the Affiliated Hospital of Xuzhou Medical University because of a cough and blood-stained sputum for longer than 1 month. Chest computed tomography (CT) suggested malignancy (Figure 1(a–c)). Positron emission -CT showed massive high-density imaging in the lower lobe of the right lung, with a maximum cross-section of 76 × 58 mm, and increased fluorodeoxyglucose metabolism (maximum standard unit value: 13.8 g/mL). The lesion margin was lobed, with a high-density shadow in the bronchus of the lower lobe of the right lung, distal occlusion, lymph node shadow of enlargement, and increased metabolism under the carina. No considerable signs of effusion were detected on either side of the chest (Figure 1(d)). After completing the relevant examination, thoracoscopic-assisted right lower lobectomy with lymph node dissection was performed.

Preoperative imaging examinations. (a–c) Chest computed tomography shows the possibility of malignancy and (d) positron emission tomography-computed tomography shows massive high-density imaging in the lower lobe of the right lung, with a maximum cross-section of 76 × 58 mm, and increased fluorodeoxyglucose metabolism (maximum standard unit value: 13.8 g/mL).
One month after the surgery, chest CT showed right pleural effusion and multiple nodules in the left lung, which strongly suggested metastasis. The patient received docetaxel and cisplatin chemotherapy for a total of six courses, and any adverse reactions were mild. The patient did not develop cough, sputum, or blood-stained sputum. After chemotherapy, the cancer antigen 125 concentration in peripheral blood was slightly increased, and there were no major abnormalities in a blood routine test, renal function, liver function, or other tumor markers. CT showed effective absorption of pleural effusion, and a left lobe pulmonary nodule was not detected (Figure 2).

One month after surgery (19 January 2016), chest computed tomography shows right pleural effusion and multiple nodules in the left lung. After treatment, the pleural effusion was gradually absorbed. After six courses of chemotherapy (9 August 2016), computed tomography shows effective absorption of pleural effusion, and a left lobe pulmonary nodule was not detected.
Pathological features
In a gross examination, the size of the surgically removed lung tissue was 15.5 × 13 ×7 cm. At a distance of 3.8 cm from the pulmonary hilum, a nodular mass with a size of 6.8 × 5.5 × 5 cm was observed under the pulmonary capsule. The tissue was grayish-red and soft with obvious necrosis.
In a microscopic examination, epithelial components and mesenchymal differentiation components were observed in tumor tissue. The epithelial component consisted of well-differentiated fetal adenocarcinoma. The single nucleolus and cytoplasmic transparent pseudo-lamellar columnar cells formed branched tubules. Mulberry bodies and neuroendocrine cells were observed (Figure 3). The mesenchymal differentiation components comprised short spindle cells, with high nuclear/plasma ratios and sporadic mitosis (Figure S1). Combined with the immunohistochemical results, the pathological diagnosis was PB. There was one lymph node metastasis in the superior paratracheal lymph nodes, and two sub-protuberant lymph nodes had metastasis.
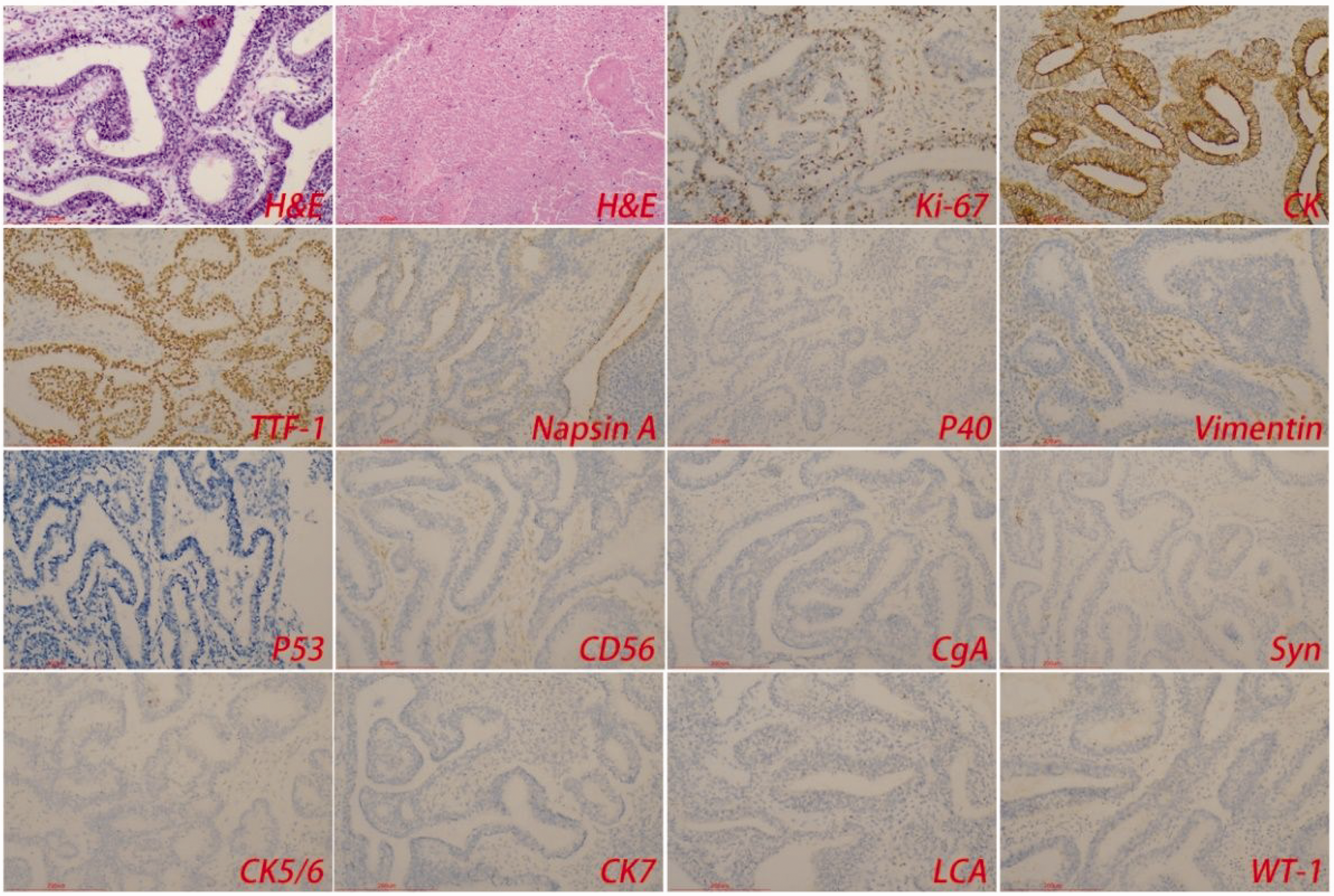
Hematoxylin and eosin and immunohistochemical staining of epithelial components. The epithelial component consists of well-differentiated fetal adenocarcinoma. A single nucleolus and cytoplasmic transparent pseudolamellar columnar cells form branched tubules. Local necrosis was observed. Tumor cells express cytokeratin and thyroid transcription factor-1.
The immunohistochemical results were as follows. Epithelial components comprised Ki-67 (+40%), TTF-1 (+), CD56 (−), Syn (−), CgA (−), leukocyte common antigen (−), P40 (−), CK5/6 (−), CK7 (−), napsin A (+), vimentin (−), CK (+), Wilms tumor 1 (−), calretinin (−), and D2-40 (−). Mesenchymal differentiation components were Ki-67 (+80%), TTF-1 (−), CD56 (+), Syn (−), CgA (−), leukocyte common antigen (−), P40 (−), CK5/6 (−), CK7 (−), napsin A (−), vimentin (focal +), CK (−), WT-1 (−), calretinin (−), and D2-40 (−) (Figure 3 and Figure S1).
Related indicators of targeted therapy
Epithelial components of related indicators of targeted therapy were programmed cell death protein 1 (−), programmed death-ligand 1 (+<1%), human epidermal growth factor receptor-2 (HER-2) (0), MLH1 (+), MSH2 (+), and MSH6 (+) (Figure S2). Mesenchymal differentiation components were programmed cell death protein 1 (−), programmed death-ligand 1 (+<1%), HER-2 (0), MLH1 (+), MSH2 (+), and MSH6 (+) (Figure S3). These results indicated that the efficacy of immunotherapy and HER-2-targeted therapy may not be ideal, while chemotherapy drugs may have a good therapeutic effect.
Gene sequencing showed that PB in this case had gene mutations of BRCA2 (exon 27) and MET (exon 15). No mutations were detected at other sites (e.g., KRAS, MAP2K1, MTOR, MYC, NRAS, NTRK1, NTRK2, NTRK3, PDGFRA, and PIK3CA). If necessary, drugs targeting these sequences may be considered in drug selection.
Follow-up visits
Chest CT was performed at 11, 15, and, 20 months, and at 2 , 3, 4, 5, 6, and 7 years after surgery, and no recurrence was found (Figure S4).
Discussion
PB is an extremely rare malignant tumor, which often occurs with distant metastasis and recurrence. The prognosis of these patients is poor, and it is related to the TNM stage, bidirectional histological type, and recurrence. 7 The incidence of metastasis is highest within 2 years of initial treatment. In 1945, Barnett and Barnard first described PB and referred to it as “embryonoma.”8,9 The pathogenesis of PB is currently unknown, and the origin of the biphasic cell population remains elusive, but is possibly related to p53 gene mutation and overexpression. The mutation of the p53 gene includes the benzopyrene gene associated with smoking, which indirectly indicates that p53 mutation may be a molecular biological mechanism of PB induced by smoking. 10 The clinical symptoms and imaging findings of PB are similar to those of non-small cell lung cancer, and it is extremely easy to be misdiagnosed clinically. Generally, PB occurs at a relatively young age and is usually a single lesion, and hilar and mediastinal lymph node metastases are less common. At present, the treatment of PB is mainly surgical resection. Lobectomy and lymph node dissection are usually performed as routine procedures for PB. The use of chemoradiotherapy for PB is controversial. Some scholars believe that postoperative radiotherapy does not improve the survival rate, but radiotherapy is feasible if there is lymph node metastasis or surrounding tissue invasion. Some studies have suggested that patients with early PB do not benefit from postoperative chemotherapy.11–14 Our study showed that postoperative use of docetaxel and cisplatin effectively treated PB recurrence and prolonged the patient’s survival time. Therefore, establishing a clinical diagnosis and treatment guidelines for PB through multi-center collaboration has important scientific value.
The 5th World Health Organization classification of thoracic tumors proposes that PB is a distinct form of lung sarcomatoid carcinoma. The most common general presentation of PB is a nodular mass, 1 to 20 cm in diameter, with clear tumor boundaries, no envelope, grayish-brown sections, and necrotic and bleeding areas. Microscopically, the epithelial components of PB are similar to those of the 11- to 18-week fetal lung, with branched or back-to-back glandular hyperplasia, columnar cells, a smaller cytoplasm, small, uniform, round to oval nuclei, and less atypia. Morula bodies may be found in 40% of PB cases. 3 Mesenchymal differentiation components consist of primitive oval or spindle cells. Allogenic differentiation, including skeletal muscle, bone, and cartilage, is observed in 25% of cases. Nuclear polymorphism and nuclear fissions are occasionally observed. Adenocarcinoma or poorly differentiated carcinoma areas can also be observed in 30% to 50% of patients with PB.15,16
A preoperative pathological diagnosis is difficult and puncture specimens are easily misdiagnosed because of the complex tissue composition of PB. 17 In cytology, there is dimorphism of epithelial cells and mesenchymal cells, and the proportion of the two compartments is different in different cases, which has a certain correlation with the prognosis of these patients. 18 Immunohistochemical staining is helpful for the pathological diagnosis and differential diagnosis of PB. Tumor cells in the epithelial component are positive for keratin staining (e.g., CAM5.2 and CK). Epithelial membrane antigen and TTF-1 are also positive. Neuroendocrine cells are scattered throughout the tumor, and they express CD56, Syn, or CgA. β-catenin staining of glands and morula bodies is nucleo-positive. Vimentin staining of mesenchymal differentiation components is positive, and other more specific markers may be displayed in heterodifferentiated regions. These immunohistochemical findings are consistent with those in our case.
A CTNNB1 exon 3 missense mutation is often present in PB, which leads to activation of the Wnt pathway through abnormal nuclear localization of β-catenin protein. TP53 mutations can also occur in PB. Mutations in ROS1 and EGFR may co-occur with mutations in CTNNB1. DICER1 and CTNNB1 mutations are present in some cases of PB, which may have a potential genetic link to pleuropulmonary blastoma in children.17,19,20 PB in our case showed gene mutations of BRCA2 and MET. If necessary, drugs targeting these sequences may be considered when selecting treatment drugs. Pathologically, PB needs to be distinguished from fetal lung adenocarcinoma, pleuropulmonary blastoma, bidirectional synovial sarcoma, and certain metastatic tumors (especially malignant mixed Miller tumors of female reproductive origin). Fetal adenocarcinoma lacks the mesenchymal embryonic cell component. Bidirectional synovial sarcoma lacks the features of fetal lung adenocarcinoma. Pleuropulmonary blastoma often occurs in children and can form a sac lined with normal respiratory epithelium.21,22
Conclusion
PB is a rare malignant lung tumor, and surgical resection and postoperative chemoradiotherapy are the preferred treatments. We report an unusual case of PB that showed a considerable therapeutic response to docetaxel and cisplatin. Our report provides new ideas and treatment options for the diagnosis and treatment of PB.
The reporting of this study conforms to the CARE guidelines. 23
Supplemental Material
sj-pdf-1-imr-10.1177_03000605241254778 - Supplemental material for Pulmonary blastoma with a good prognosis: a case report and review of the literature
Supplemental material, sj-pdf-1-imr-10.1177_03000605241254778 for Pulmonary blastoma with a good prognosis: a case report and review of the literature by Qingru Liu, Fengchao Li, Lulu Wang, Feng Lv, Qianqian Yin, Guangzhen Liu and Wen Chen in Journal of International Medical Research
Footnotes
Acknowledgements
The authors thank the patient for her cooperation and permission to publish this case.
Author contributions
Q. L. wrote the manuscript and collected the data. Fc. L, L. W, F. L, Q. Y, and G. L prepared the figures. W. C. contributed to the data analysis and helped prepare all figures and tables, and edited the manuscript. All authors reviewed the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Ethics statement
This study was approved by the medical ethics committee of the Affiliated Hospital of Xuzhou Medical University (approval number: xyfy202254) and was in accordance with the Helsinki Declaration of 1975. The patient provided written consent for publication of this report.
Funding
This work was supported by the National Natural Science Foundation of China (Nos: 31800814, 32271411).
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.