
Abstract
Objective
To construct a prognostic model of a breast cancer-related oxidative stress-related gene (OSRG) signature using machine learning algorithms.
Methods
The OSRGs of breast cancer were constructed by least absolute shrinkage and selection operator (LASSO) and multivariate Cox regression analysis. The Cancer Genome Atlas (TCGA) was used to analyse the gene expression and prognostic value. The Human Protein Atlas was used to analyse the protein expression of hub genes. Receiver operating characteristic analysis, calibration curve and decision curve analysis were used to predict the stability of this model.
Results
The area under the curve of 1-, 3- and 5-year overall survival were 0.751, 0.707 and 0.645 in the TCGA training dataset; and 0.692, 0.678 and 0.602 in the TCGA testing dataset, respectively. Calibration plot showed good agreement between predicted probabilities and observed outcomes. Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Set Enrichment Analysis (GSEA) pathway analysis indicated that multiple cancer-related pathways were highly enriched in the high-risk group. Immune infiltration analysis showed immune cells and their functions may play a key role in the development and mechanism of breast cancer.
Conclusions
This new OSRG signature was associated with the immune infiltration and it might be useful in predicting the prognosis in patients with breast cancer.
Introduction
Breast cancer continues to rank as the foremost threat to female health globally, necessitating urgent attention towards active and effective treatment and prevention strategies. Over the past two decades, significant breakthroughs have been achieved in human research pertaining to the treatment and pathogenesis of breast cancer. 1 One of the gravest consequences of breast cancer is its propensity to metastasize to vital organs such as the liver, brain, lungs and bones during the middle and late stages, leading to the deterioration of these target organs, impairment of their function and ultimately resulting in the demise of patients. 2 Bone metastases are particularly prevalent in breast cancer, disrupting skeletal homeostasis and initiating a detrimental cycle. 3 A previous report highlighted that brain metastases from breast cancer are frequently observed in cases of human epidermal growth factor receptor 2 (HER2)-positive or triple-negative breast cancers. 4 The intricate interaction between cancer cells and the microenvironment of target organs is crucial, with circulating tumour cells engaging in interactions with brain astrocytes to activate complex signalling pathways, including PI3K-AKT, HER2-HER3 signalling, among others. 4
In recent years, there has been a growing emphasis on exploring the role of oxidative stress in the pathogenesis of tumours. Numerous studies have highlighted the presence and progression of oxidative stress in breast cancer. Specifically, investigations have revealed that the overexpression of the glutamine transporter SNAT2/SLC38A2 in triple-negative breast cancer significantly augments resistance to oxidative stress, thereby contributing to an unfavourable prognosis in breast cancer cases. 5 The reactive oxygen species generated during episodes of oxidative stress exert notable effects on various signalling pathways within the human body. These alterations can compromise the body’s innate defence mechanisms against tumour angiogenesis and the metastasis of cancer cells, emerging as a pivotal factor in the advancement of breast cancer. 6
Oxidative stress gene prognostic signatures are currently being increasingly studied in some cancers. An in-depth study investigated three oxidative stress gene signatures in ovarian cancer patients receiving platinum-based chemotherapy. 7 A four oxidative stress gene signature was constructed to predict survival in clear cell renal cell carcinoma. 8 With the escalating prominence of oxidative stress in breast cancer research, the exploration of prognostic factors associated with oxidative stress in the development and progression of breast cancer has emerged as a focal point of investigation. A previous study constructed an oxidative stress long noncoding RNA prognostic signature for breast cancer and proved that it is closely related to the tumour microenvironment. 9 Another study successfully constructed a clinical prognostic gene signature for triple-negative breast cancer. 10 However, the current state of research on prognostic genes related to oxidative stress in breast cancer, and whether these genes can serve as viable biomarkers for predicting breast cancer prognosis, remains ambiguous. This warrants further exploration. This current study, using the TCGA database and pertinent oxidative stress genes documented in the existing literature, employed bioinformatics and other methodologies to construct a prognostic gene model specific to oxidative stress in breast cancer. This initiative aims to offer novel perspectives on both the prognosis and underlying biological mechanisms of breast cancer.
Materials and methods
Ethical statement
This current study used bioinformatics analysis of data available in public databases. Therefore, local ethical committee approval and informed consent were not required.
Breast cancer expression profile data, clinical data download and acquisition of oxidative stress-related genes
Gene expression profiling, clinicopathological and prognostic data concerning breast cancer patients were obtained from the Breast Invasive Carcinoma (BRCA) Project RNAseq data available in the HTSeq-FPKM format, Level 3, through the TCGA database website (https://portal.gdc.cancer.gov/). The raw data in fragments per kilobase per million (FPKM) format underwent conversion to transcripts per million reads (TPM) format, followed by a log2 transformation. Upon thorough inspection, the dataset was confirmed to be complete. A compilation of 1076 genes associated with oxidative stress was curated from the Gene Cards database (https://www.genecards.org) or derived from the “GO_RESPONSE_TO_OXIDATIVE_STRESS” dataset acquired from the Gene Set Enrichment Analysis (GSEA) website (https://www.gsea-msigdb.org/gsea/datasets.jsp). The differential expression analysis involved the extraction of genes from 1100 breast cancer samples using the limma package, with criteria set at |log2 fold-change [FC]| > 0.5 and P < 0.05, signifying statistical significance. 11
GO and KEGG enrichment analysis and PPI net construction
The study employed a Venn diagram to identify differentially expressed oxidative stress-related genes (DE-OSRGs) by intersecting differentially expressed genes and oxidative stress genes. The analysis utilized the Cluster Profiler package (version 3.14.3) for Gene Ontology (GO) and KEGG enrichment analysis. Additionally, the Hs.eg.db package (version 3.10.0) was used for ID conversion.12,13 GO categorizes biological information into cellular component (CC), molecular function (MF) and biological process (BP). By employing GO analysis, the current analysis elucidated the primary functions associated with the differential genes across these three levels: CC, MF and BP. KEGG enrichment analysis was instrumental in identifying the signalling pathways predominantly enriched by the DE-OSRs. For Protein–Protein Interaction (PPI) Enrichment Analysis, Metascape (http://metascape.org/) served as the chosen tool.
Correlation analysis between oxidative stress-related genes and cancer related pathways
RNA-sequencing expression (level 3) profiles and corresponding clinical information for BRCA were downloaded from the TCGA dataset (https://portal.gdc.com). The R software GSVA package was used to analyse, choosing parameter as method=‘ssgsea’. The correlation between genes and pathway scores was analysed by Spearman’s correlation. All the analysis methods and R packages were implemented by R version 4.0.3. A P-value < 0.05 was considered statistically significant.
Gene set enrichment analysis
For gene set enrichment analysis (GSEA), a predefined set of differentially expressed genes, sourced from the MsigDB database, was employed to arrange the log fold change (log FC) in descending order. This ordering serves to illustrate the directional trend in gene expression changes between the two groups. The upper portion of the list signifies upregulated differential genes, while the lower portion denotes downregulated differential genes.
Construction of prognosis model of oxidation stress genes
The R package was used for the execution of univariate Cox regression analysis on differentially expressed oxidative stress genes to discern prognostic factors. Subsequently, all patients underwent random allocation into training and testing groups. The identified genes were then subjected to least absolute shrinkage and selection operator (LASSO) regression and univariate Cox regression analysis, facilitating the construction of a set of core genes associated with oxidative stress. Ultimately, patients were stratified into high- and low-risk groups based on the median risk score. Survival analysis, receiver operating characteristic (ROC) analysis, calibration curve analysis and decision curve analysis were performed on both the training and testing groups to evaluate the robustness of the model.
The LASSO regression model was used for the selection of genes to identify optimal gene sets associated with survival outcomes. The formula for the LASSO algorithm is as follows:
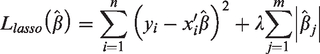
Immunohistochemical analysis
The protein expression levels within the hub genes of the DE-OSRGs model were examined through the Human Protein Atlas (HPA) database (https://www.proteinatlas.org/).
Immunocyte infiltration analysis
The TRNA-sequencing expression profiles (level 3) alongside corresponding clinical data for breast cancer were obtained from the TCGA dataset accessible at https://portal.gdc.com. Gene expression immune scores correlations were computed employing the ggstatsplot package. Pearson correlations were employed for normally distributed data, while Spearman’s correlations were used for data that were not normally distributed. Statistical significance was acknowledged for two-sided P-values <0.05.
Results
TCGA-BRCA data download and baseline data included in the population
The flow chart of this study is presented in Figure 1. The dataset included a total of 1110 breast cancer patients for analysis in this study. Pertinent clinical, demographic and pathological characteristics have been documented in Table 1. The overlap between TCGA breast cancer differential genes and oxidative stress genes is depicted in the Venn diagram (Figures 2a & 2b). GO and KEGG enrichment analyses revealed that differentially expressed oxidative stress genes in breast cancer exhibited predominant enrichment in biological processes such as cellular oxidative stress, reactive oxygen species metabolic processes and responses to reactive oxygen species (BP). Additionally, they demonstrated enrichment in cellular components, including membrane raft, blood microparticle and secretory granule lumen (CC); as well as molecular functions, encompassing growth factor binding, cytokine receptor binding and cytokine activity (MF) (Figure 2c). The KEGG analysis highlighted significant enrichment in pathways like proteoglycans in cancer and the tumour necrosis factor signalling pathway. The results of the PPI analysis are presented in Figure 2d.

Flow chart of the overview of this study design. RNA Seq, RNA sequence; BC, breast cancer; TCGA, The Cancer Genome Atlas; OSRGs, oxidative stress-related gene; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, Protein–Protein Interaction; DE-OSRGs, differentially expressed oxidative stress-related genes; LASSO, least absolute shrinkage and selection operator; OS, oxidative stress; ROC, receiver operating characteristic; DCA, Decision Curve Analysis; HPA, Human Protein Atlas; GSEA, Gene Set Enrichment Analysis.
The demographic and clinical characteristics of the patients (n = 1100) with breast cancer that were included in The Cancer Genome Atlas database.

Data only available for 1090 patients.
IQR, interquartile range; T, tumour; N, node; M, metastasis; OS, overall survival.

Acquisition and functional enrichment analysis of differentially expressed oxidative stress genes. Venn diagram of differentially expressed oxidative stress genes (a). Volcano diagram of DE-OSRGs in breast cancer (b). GO and KEGG enrichment analysis of DE-OSRGs in breast cancer (c). PPI analysis of DE-OSRGs in breast cancer (d). DE-OSRGs, differentially expressed oxidative stress-related genes; GO, Gene Ontology; BP, biological process; CC, cellular component; MF, molecular function; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, Protein–Protein Interaction; TNF, tumour necrosis factor; AGE-RAGE, advanced glycation end products-receptor for advanced glycation end products; MAPK, mitogen-activated protein kinase. The colour version of this figure is available at: http://imr.sagepub.com.
The GSEA highlighted the enrichment of the investigated gene in various pathways (Figure 3). Specifically, the gene demonstrates enrichment in the calcium signalling pathway (A), the P53 signalling pathway (B), the FOXM1 signalling pathway (C), biological oxidations (D), TP53 activity through phosphorylation € and signalling by receptor tyrosine kinases (F). The OR-related core genes (MAP2K6, CACNA2D1, SDC1) are mainly concentrated in cancer-related pathways such as tumour inflammation, cell proliferation, apoptosis, DNA repair and angiogenesis (see supplementary materials, Figures S1–S3).

Gene Set Enrichment Analysis (GSEA) results of differential genes. The GSEA analysis indicates that this gene is enriched in KEGG_CALCIUM_SIGNALING_PATHWAY (a), KEGG_P53_SIGNALING_PATHWAY (b), PID_FOXM1_PATHWAY (c), REACTOME_BIOLOGICAL_OXIDATIONS (d), REACTOME_REGULATION_OF_TP53_ACTIVITY_THROUGH_PHOSPHORYLATION (e) and REACTOME_SIGNALING_BY_RECEPTOR_TYROSINE_KINASES (f). The colour version of this figure is available at: http://imr.sagepub.com.
Constructing the OSRG model in breast cancer using LASSO regression
The TCGA dataset was randomly divided into training and testing datasets at a ratio of 6:4. Subsequently, patients within these datasets were stratified into high-risk and low-risk categories based on the median risk score. Univariate and multivariate Cox analyses were conducted in both the training and testing groups (Table 2). A risk score model was ultimately constructed to determine the optimal coefficients, utilizing the LASSO regression method (Figures 4a & 4b). The LASSO regression results confirmed CACNA2D1 and MAP2K6 as protective factors for breast cancer, while SDC1 was a risk factor for breast cancer (Figure 4c). The survival analysis using Kaplan–Meier curves demonstrated the robust stability of the OSRGs signature in predicting the prognosis of breast cancer (Figures 4d & 4e). The risk score was calculated using the following equation: risk score = (−0.164*CACNA2D1) + (−0.225*MAP2K6) + (0.247*SDC1).
Univariate and multivariate Cox analysis in the training and testing groups.

HR, hazard ratio; CI, confidence interval; NS, not significant (P ≥ 0.05).

Least absolute shrinkage and selection operator (LASSO) regression analysis was selected to construct the prognostic model. Screen of Partial Likelihood Deviance and the best Lambda (a). Excellent coefficients of each gene were obtained by optimizing Lambda (b). The hazard ratio result of the LASSO regression for each hub gene (c). The overall survival of differentially expressed oxidative stress-related genes (DE-OSRGs) prognostic model in the training group (d). The overall survival of DE-OSRGs prognostic model in the testing group (e). The colour version of this figure is available at: http://imr.sagepub.com.
The Kaplan–Meier curves (Figures 5a & 5b) revealed a noteworthy disparity in overall survival (OS) between the high-risk score group and the low-risk score group. Specifically, the OS duration in the high-risk score group was significantly reduced compared with that in the low-risk score group.
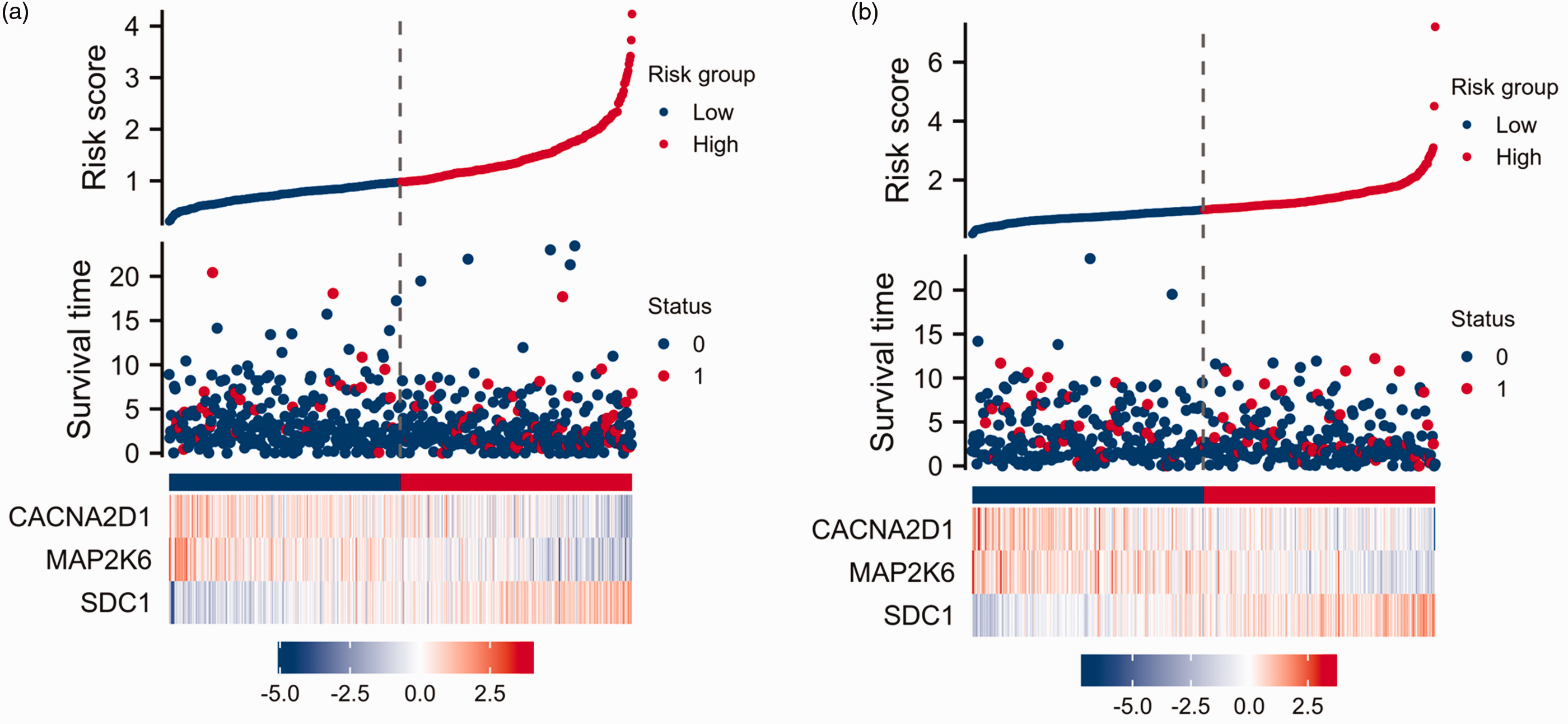
Construction of risk score in the training group (a) and testing group (b). The colour version of this figure is available at: http://imr.sagepub.com.
The expression of hub genes and validation of the protein levels using the HPA database
The CACNA2D1 protein did not display differential regulation between BC and normal tissues, which contradicts the CACNA2D1 gene expression pattern observed (Figures 6a & 6b). Further experiments should be undertaken to elucidate this discrepancy. The expression of the MAP2K6 protein in BC tissues was lower compared with normal tissues, which conforms with the gene expression (Figures 6d & 6e). The expression of the SDC1 protein was notably higher in BC tissues than in normal tissues, consistent with the gene expression profile (Figures 6g & 6h). All three core genes have significant prognostic value in breast cancer (Figures 6c, 6f & 6i).

Analysis of every hub gene at the protein or gene level and overall survival in breast cancer. The expression of CACNA2D1 gene (a) or CACNA2D1 protein (b) and result of overall survival in breast cancer (c). The expression of MAP2K6 gene (d) or MAP2K6 protein (e) and result of overall survival in breast cancer (f). The expression of SDC1 gene (g) or SDC1 protein (h) and result of overall survival in breast cancer (i). The colour version of this figure is available at: http://imr.sagepub.com.
The verification of robustness of the prognosis model
To enhance the prognostic predictive capability of this signature in breast cancer (BC) patients, we performed an overall survival prediction at 1-year, 3-year, and 5-year intervals using the ROC method. The ROC analysis results demonstrated a good predictive ability at 1-year and 3-year intervals (training dataset: AUC = 0.751, 0.707, respectively; testing dataset: AUC = 0.692, 0.678, respectively) compared with the 5-year interval (training dataset: AUC = 0.645; testing dataset: AUC = 0.602; Figures 7a & 7b). Furthermore, the calibration curve illustrated a consistent performance between the training dataset and testing dataset (Figures 7c & 7d). Subsequently, a Decision Curve Analysis was undertaken to assess the net benefit of various parameters. The findings indicated that the combination of the ‘signature’ and ‘Tumour, Node, Metastasis’ achieved an optimal net benefit in the prediction of 3-year OS (Figures 7e & 7f).

Validation of differentially expressed oxidative stress-related genes (DE-OSRGs) prognostic model. Receiver operating characteristic (ROC) analysis of DE-OSRGs signature in the training (a) and testing groups (b). Calibration curve analysis of DE-OSRGs signature in the training (c) and testing groups (d). Decision Curve Analysis of DE-OSRGs signature in the training (e) and testing group (f). TCGA, The Cancer Genome Atlas; TPR, true positive rate; FPR, false positive rate. The colour version of this figure is available at: http://imr.sagepub.com.
The association between OSRGs and tumour immune cells
Oxidative stress genes frequently exhibit close associations with tumour immune cells and the microenvironment. An analysis was undertaken to assess the correlations between the risk score associated with this signature and the infiltration of immune cells within the tumour. The results revealed a pronounced negative correlation, indicating that the risk score was inversely associated with the expression levels of T cell
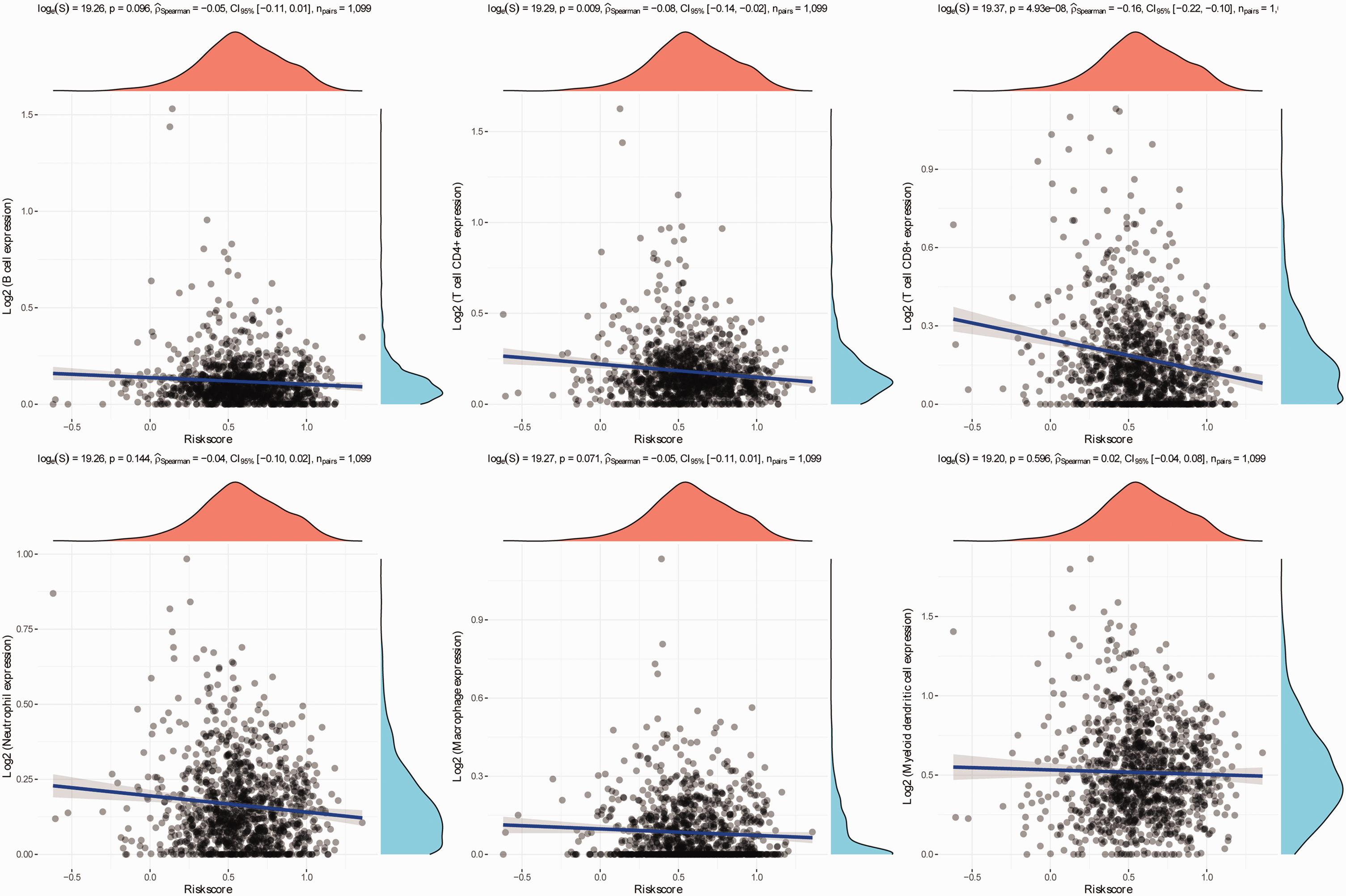
Correlation analysis between risk scores of oxidative stress gene model and immune cells. The colour version of this figure is available at: http://imr.sagepub.com.
Discussion
Breast cancer has emerged as a global health concern, representing the most prevalent form of cancer affecting women. 1 Scholars worldwide have expressed considerable interest in both preventing and treating breast cancer patients. Extensive research has consistently affirmed the disruption in the equilibrium between pro-oxidation and antioxidants.14,15 The generation of reactive oxygen species (ROS) and the induction of oxidative stress are recognized as factors associated with the onset and progression of breast cancer.14,16 An excess production of ROS or an inadequacy in the antioxidant system precipitates an imbalance in oxidation and antioxidants. This imbalance, in turn, instigates DNA damage and mutations in tumour suppressor genes, thereby serving as the catalyst for the initiation of breast cancer. 17 There are currently many reports on anti-oxidative stress genes, such as nuclear factor E2-related factor 2-antioxidant response element (Nrf2-ARE), glutathione reductase (GR), superoxide dismutase (SOD), heat shock protein (HSP) and FOXO proteins.18–20 Nrf2 is a transcription factor and when cells are subjected to oxidative stress, Nrf2 is transferred from the cytoplasm to the nucleus and binds to antioxidant response element to promote the transcription of a series of antioxidant genes, such as the GR and SOD genes, which enhances the antioxidant capacity of cells. 21 Under oxidative stress conditions, FOXO protein activity is activated and promotes the expression of a series of antioxidant response genes, such as SOD and glutathione peroxidase (GPX). 22 In breast cancer cells, mitochondrial signal transducer and activator of transcription 3 plays a role in regulating cellular redox balance by regulating antioxidant gene expression. 23 Research has shown that breast cancer type 1 susceptibility protein (BRCA1) can decrease ROS levels, stimulate antioxidant gene expression and protect cells from oxidative stress in breast cancer. 24 These findings suggest that antioxidant-related genes play a crucial role in antioxidant systems in breast cancer.
In recent years, there has been a growing focus on investigating the expression and functionality of oxidative stress genes across various tumour types. Specifically, scholars have undertaken evaluations concerning the association between 11 single nucleotide polymorphisms within oxidative stress genes, MT2A, NFE2L2, NQO1, PRDX1 and PRDX6, and overall breast cancer mortality. Previous findings have indicated a significant correlation between these gene polymorphisms and total breast cancer mortality. 25 Simultaneously, these oxidative stress-related genes exhibit close interactions with tumour immune cells and the tumour microenvironment. 26 In the context of oestrogen receptor (ER)-positive breast cancer, oxidative stress genes can influence the invasion and growth of ER-positive breast cancer cells through their interaction with oestrogen receptors. 27 Furthermore, the sustained presence of oxidative stress may lead to alterations in metabolic pathways within certain tumour cells. ROS produced in breast cancer have been implicated in fostering resistance to therapy and inducing cell apoptosis. 15 Collectively, these investigations consistently affirm a robust association between oxidative stress genes and the development of breast cancer.
In previous investigations, the prognostic implications of oxidative stress genes have been extensively documented across various malignancies, although scant attention has been directed toward their role in breast cancer.7,9,14,28 To address this gap in knowledge, this current study undertook a comprehensive bioinformatic analysis, yielding a novel and meaningful oxidative stress gene signature for breast cancer. Earlier research had demonstrated the upregulation of CACNA2D1 in several cancers, including lung cancer, ovarian cancer, and gastric cancer, correlating with an unfavourable prognosis.29,30 Intriguingly, the paucity of experimental investigations into the role of CACNA2D1 in breast cancer should be worthy of in-depth study. Hence, the analysis of the CACNA2D1 gene is worthy of attention in the future. The current study demonstrated that the levels of CACNA2D1 gene expression were not consistent with the protein levels, with CACNA2D1 protein not showing differential regulation between BC and normal tissues. There might be several reasons for this phenomenon. First, gene expression is divided into two phases: transcription and translation, which are characterized by the production of mRNA and protein, respectively. The time and site of transcription and translation of eukaryotic gene expression are spatiotemporal. Secondly, after transcription, there are several levels of post-transcription processing, degradation of transcription products, translation, post-translation processing and modification. So, the levels of transcription and translation are not the same. Thirdly, due to the different time-points of detection, the mRNA may have been degraded when the protein reached its peak, or the protein amount may still be increasing when the mRNA reached its peak. Notably, MAP2K6 assumes a pivotal role in the progression of multiple cancers, such as the regulation of gastric cancer development via the miR-1298-5p/MAP2K6/p38 MAPK axis. 31 Recent findings corroborate the prognostic prowess of a 16-gene signature involving MAP2K6 in breast cancer patients, further emphasizing its significance. 32 Additionally, SDC1 emerges as an independent prognostic risk factor associated with poor overall survival in breast cancer, 33 reinforcing the implications of the current findings. Collectively, these observations underscore the substantial value of the MAP2K6 gene in the model that was constructed in the current study.
Function annotation analyses were conducted to investigate the mechanisms underlying the differential expression of oxidative stress-related genes between groups with high- and low-risk scores. The current findings revealed significant involvement of proteoglycans in cancer, the MAPK signalling pathway, FOXM1 and the P53 signalling pathway, all of which typically play crucial roles in breast cancer development.34–36 In summary, the current study indicates that oxidative stress-related genes may play a role in the development of breast cancer.
The current study addresses the important issue of breast cancer by examining the predictive role of oxidative stress-related genes. Initially, a robust and innovative gene signature for oxidative stress in breast cancer was constructed using the LASSO regression method with data from the TCGA database. This signature aids in deepening our understanding of the onset and progression of breast cancer and in predicting its prognosis. As anticipated, the validated oxidative stress-related signature demonstrated strong predictive performance for 1-year, 3-year and 5-year OS, with particular efficacy in the 1-year and 3-year periods. While previous studies have developed signatures for breast cancer-related genes, models specifically focusing on oxidative stress-related genes are infrequently reported. Then a thorough analysis of the expression of hub genes using data from the TCGA and HPA databases was undertaken. Finally, a comprehensive clinical prediction model was established to validate this signature in predicting the prognosis of breast cancer patients. There is ample evidence to demonstrate the stability of this model.
This study had several limitations. First, it did not include biological experiments to validate the obtained results. In future, incorporating more detailed experiments would enhance the robustness and credibility of the current findings. Additionally, the inclusion of more diverse clinical datasets is imperative to ensure the generalizability of the current conclusions. Secondly, the study was constrained by a limited sample size, which curtailed the scope of further verification. To address this limitation and bolster the statistical power of the analysis, there is a plan to undertake a comprehensive large-scale, multicentre study in subsequent research endeavours.
In conclusion, this current study has developed a signature comprising three oxidative stress-related genes. This signature demonstrates robust prognostic value in predicting outcomes among breast cancer patients. Moreover, its integration with other clinical parameters yields enhanced clinical net benefits.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605241232560 - Supplemental material for Construction of an oxidative stress-associated genes signature in breast cancer by machine learning algorithms
Supplemental material, sj-pdf-1-imr-10.1177_03000605241232560 for Construction of an oxidative stress-associated genes signature in breast cancer by machine learning algorithms by Daojun Hu, Bing Qin, Li Zhang and Hanli Bu in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605241232560 - Supplemental material for Construction of an oxidative stress-associated genes signature in breast cancer by machine learning algorithms
Supplemental material, sj-pdf-2-imr-10.1177_03000605241232560 for Construction of an oxidative stress-associated genes signature in breast cancer by machine learning algorithms by Daojun Hu, Bing Qin, Li Zhang and Hanli Bu in Journal of International Medical Research
Supplemental Material
sj-pdf-3-imr-10.1177_03000605241232560 - Supplemental material for Construction of an oxidative stress-associated genes signature in breast cancer by machine learning algorithms
Supplemental material, sj-pdf-3-imr-10.1177_03000605241232560 for Construction of an oxidative stress-associated genes signature in breast cancer by machine learning algorithms by Daojun Hu, Bing Qin, Li Zhang and Hanli Bu in Journal of International Medical Research
Footnotes
Author contributions
Daojun Hu and Hanli Bu collectively wrote this manuscript. Daojun Hu and Bing Qin were responsible for creating all of the figures and tables. Hanli Bu and Li Zhang contributed to the study design and provided overall supervision.
Data availability statement
All the datasets provided in this current study can be obtained in TCGA (https://portal.gdc.cancer.gov/), Gene Cards database (https://www.genecards.org), Gene Set Enrichment Analysis (GSEA) website (https://www.gsea-msigdb.org/gsea/datasets.jsp), Metascape (http://metascape.org/) and HPA database (![]() ).
).
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Funding
This work was supported by the Innovative and Entrepreneurial Talent program of Chongming District, Shanghai (2021).
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.