
Abstract
Ectopic adrenocorticotropic hormone syndrome (EAS) is a rare condition caused by pancreatic neuroendocrine tumors (p-NETs). The severe hypercortisolemia that characterizes EAS is associated with a poor prognosis and survival. Mitotane is the only adrenolytic drug approved by the Food and Drug Administration and is often used to treat adrenocortical carcinoma. Combination therapy with mitotane and other adrenal steroidogenesis inhibitors is common for patients with Cushing’s syndrome (CS). Here, we describe three patients who developed EAS secondary to the liver metastasis of p-NETs. All three rapidly developed hypercortisolemia but no typical features of CS. They underwent anti-tumor and mitotane therapy, which rapidly reduced their blood cortisol concentrations and ameliorated their symptoms. Their hypercortisolemia was controlled long term using a low dose of mitotane. The principal adverse effects were a slight loss of appetite and occasional dizziness, and there were no severe adverse effects. Importantly, even when the tumor progressed, the patients’ circulating cortisol concentrations remained within the normal range. In summary, the present case series suggests that mitotane could be used to treat hypercortisolemia in patients with EAS caused by advanced p-NETs, in the absence of significant adverse effects.
Keywords
Introduction
Ectopic adrenocorticotropic hormone syndrome (EAS) is a rare condition that accounts for 5% to 10% of cases of adrenocorticotropic hormone (ACTH)-dependent Cushing’s disease. 1 Malignant neoplasms that secrete ACTH include bronchial carcinoid, small-cell lung carcinoma, thymic carcinoid, pancreatic neuroendocrine tumor (p-NET), and acinic cell carcinoma of the parotid or pancreas. Of these, bronchial carcinoid is the most common cause of EAS and p-NET accounts for approximately 15% of cases.2,3 p-NET is characterized by an insidious onset and typically have already metastasized at the time of diagnosis. Advanced tumors secrete large amounts of ACTH, which causes severe acute hypercortisolemia. A high serum cortisol concentration can cause a series of complications that significantly worsen the prognosis of patients. 4 Although bilateral adrenalectomy is an effective treatment for this hypercortisolemia and prolongs the life of patients, it is associated with numerous potential intraoperative complications that increase the risk of surgical mortality. 5 Therefore, the identification of effective pharmacological treatments for hypercortisolemia would be especially important. Here, we report three patients with advanced p-NETs and EAS, whose hypercortisolemia was rapidly controlled by the administration of mitotane alone, resulting in significant clinical improvements and longer survival.
We obtained the patients’ consent for treatment, and the written informed consent for the publication of their cases was obtained from the relatives of the patients. The reporting of this study conforms to the CARE guidelines. 6
Case descriptions
Case 1
A man of approximately 40 years of age underwent abdominal surgery (distal pancreatectomy, splenectomy, partial hepatectomy, and right hemicolectomy) in April 2017 for the treatment of a pancreatic neoplasm with multiple liver metastases. During the surgery, a mass measuring 5.1 cm × 4.6 cm was found in the tail of his pancreas. Pathologic examination revealed a grade 2 (G2) neuroendocrine tumor with a Ki-67 index of 3%. Following the surgery, the patient underwent anti-tumor therapy for several years, but throughout this period, he experienced repeated episodes of hypokalemia and hyperglycemia.
In March 2019, the patient presented at our clinic for further treatment owing to tumor progression. We promptly initiated a treatment regimen comprising a combination of octreotide (sandostatin LAR) and CAPTEM (capecitabine and temozolomide). However, during the course of this treatment, he developed psychiatric symptoms, including auditory hallucination, manic episodes, and delusional episodes. Laboratory testing revealed a low blood potassium concentration of 2.4 mmol/L (normal range 3.5 to 5.5 mmol/L), high plasma ACTH concentrations of 51.6 pmol/L at 08:00 and 50.2 pmol/L at 00:00 (normal range 1.59 to 13.96 pmol/L), a high serum cortisol concentration of 1,750 nmol/L at 08:00 (normal range 118 to 868 nmol/L), and an absence of normal diurnal variation. His 24-hour urinary free cortisol production was also significantly higher than normal (Table 1). Abdominal enhanced computed tomography (CT) examination revealed hepatic tumors with irregular enhancement and bilateral hyperplasia of the adrenal glands. Magnetic resonance imaging (MRI) of his pituitary showed no abnormalities (Figure 2a, 2d, 2g).
Clinical characteristics of, and laboratory test results for, the three patients.

ACTH, adrenocorticotropic hormone; UFC, urinary free cortisol; NA, not available, AJCC, American Joint Committee on Cancer
On the basis of the patient’s medical history, symptoms, laboratory data, and imaging findings, an initial diagnosis of EAS caused by advanced p-NET was made. We subsequently proposed bilateral inferior petrosal sinus sampling to confirm the diagnosis; however, the patient declined this procedure. After a single cycle of anti-tumor treatment, the hepatic tumors exhibited significant shrinkage, and the patient’s serum ACTH and cortisol concentrations returned to normal without the necessity for any treatment for Cushing’s syndrome. The features of EAS also improved. As the tumors continued to decrease in size, the patient’s hormone concentrations and the clinical manifestations of the disease improved, providing support for the diagnosis.
Subsequently, we continued the anti-tumor therapy, which alleviated his EAS symptoms. However, his tumor started to progress again in December 2021, resulting in increases in his circulating ACTH and cortisol concentrations and a worsening of his symptoms. Therefore, we introduced a new treatment regimen for the tumor, and to manage the hypercortisolemia, we introduced mitotane at a dose of 1,000 mg three times daily. After approximately 7 days of treatment, the patient’s serum cortisol concentration had returned to normal (Figure 1), and he had not experienced any significant adverse effects. During the follow-up period, the patient’s hypercortisolemia remained under control using a maintenance dose of mitotane (serum cortisol concentration at 08:00 between 275 and 668 nmol/L), despite tumor progression. Unfortunately, the patient eventually died owing to complications of the malignancy, after surviving 66 months.
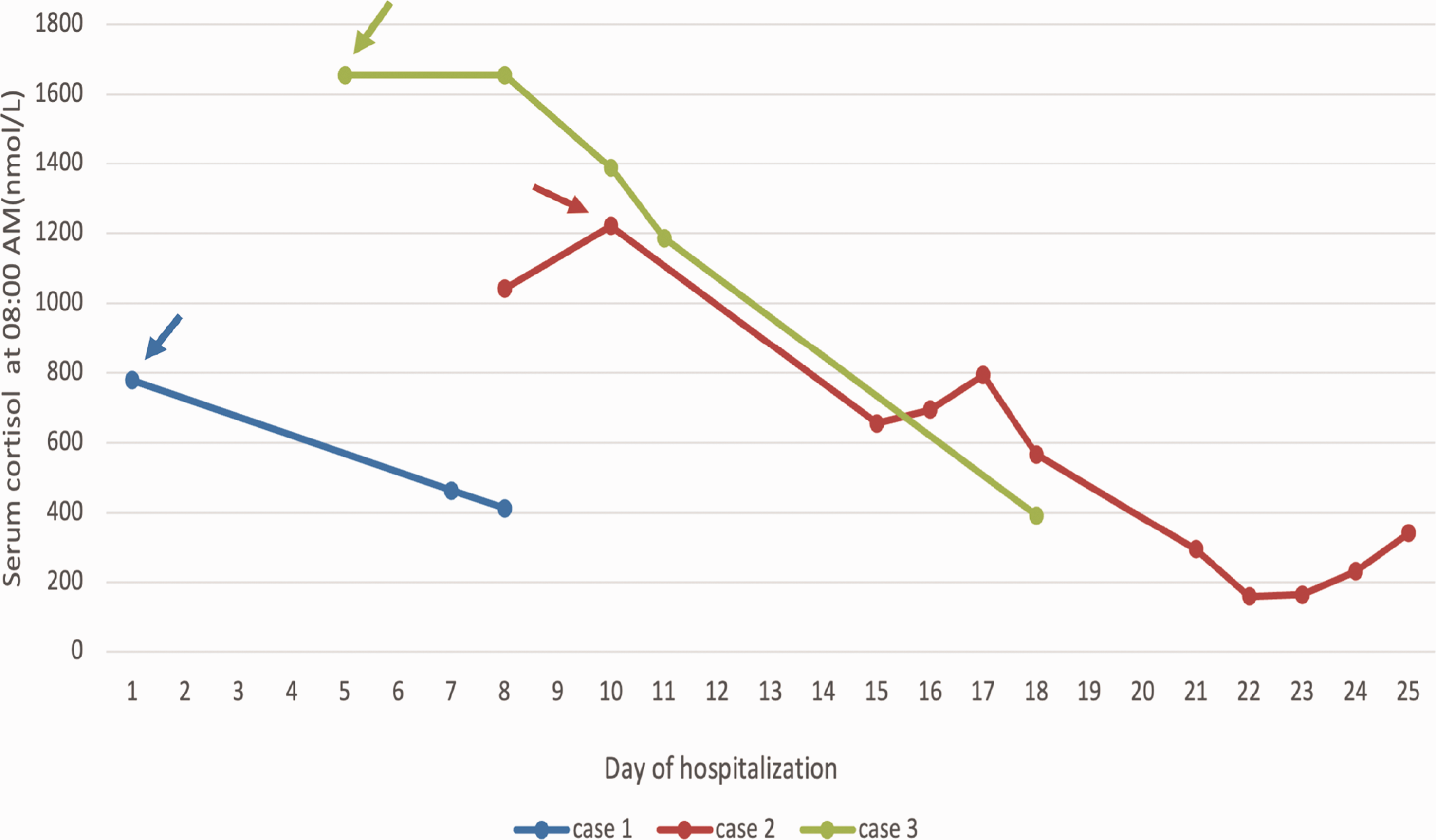
Timing of the start of mitotane administration (arrows) and the subsequent changes in the serum cortisol concentrations of the three patients.
Case 2
A man of approximately 60 years of age was admitted to his local hospital in September 2021 with fatigue and facial and limb edema. Subsequent investigations revealed the presence of a grade 2 (G2) pancreatic neuroendocrine tumor (p-NET) with multiple metastases, including in the liver, retroperitoneal lymph nodes, and bones. Following three cycles of CAPTEM chemotherapy that were not curative, the patient sought further treatment at our clinic in December 2021. During his hospitalization, he exhibited symptoms of hyperactivity, substantial appetite, and impaired sleep.
Physical examination revealed hypertension (170/80 mmHg), facial plethora, and edema of the face and limbs. However, moon face, buffalo hump, central obesity, and purple striae were absent. Laboratory testing revealed a serum potassium concentration of 3.0 mmol/L, a fasting blood glucose concentration of 23.3 mmol/L, and high plasma ACTH and serum cortisol concentrations (Table 1). Subsequently, a series of imaging examinations were performed. An abdominal enhanced CT examination revealed hepatomegaly, adrenal hyperplasia, diffuse tumors with annular enhancement in the liver, and a well-defined mass measuring 6.3 cm × 5.7 cm with regular enhancement in the tail of the pancreas, which invaded the patient’s left kidney and spleen. However, pituitary enhanced MRI did not reveal any abnormalities (Figure 2b, 2e, 2h). On the basis of these findings, we suspected that the neuroendocrine tumor might be an ectopic ACTH-secreting neoplasm.

Diagnostic images of the three patients. (a) Abdominal computed tomography (CT) showed poorly enhancing nodular lesions with unclear boundaries. (b) Abdominal CT revealed an increase in liver volume, with multiple masses showing annular enhancement. In addition, there was a mass in the tail of the pancreas that was uniformly enhancing and invaded the spleen. (c) Abdominal CT showed multiple round, low-density nodules in the liver, with mild annular enhancement. (d–f) CT showed adrenal hyperplasia (arrows) and (g–i) Pituitary enhanced magnetic resonance imaging showed that the size, shape, and signal strength of the pituitary gland were normal.
Treatment of the tumor was initiated immediately after assessing the patient’s condition using octreotide and surufatinib. In addition, mitotane administration was commenced to manage his hypercortisolemia, initially at a dose of 1,000 mg three times daily, and the patient’s serum cortisol concentration returned to normal after 6 days of this medication. Furthermore, his clinical symptoms improved. To prevent adrenal insufficiency, the patient’s circulating hormone concentrations were monitored over an extended period. Ultimately, his serum cortisol concentration remained within the normal range while administering the minimum dose of mitotane (1,000 mg/day), with occasional adjustments of the dose owing to fluctuations in hormone concentrations.
Owing to the patient’s substantial tumor burden and the high activity of the neoplasms, his plasma ACTH concentration increased to 225 pmol/L in March 2022. However, mitotane treatment was associated with stability of his serum cortisol concentration (at 08:00, ranging from 122 to 716 nmol/L) and the clinical manifestations of the disease. In addition, he experienced no significant adverse effects, with only slight loss of appetite and dizziness. Unfortunately, the patient ultimately died owing to the effects of the tumor, resulting in a duration of survival of 11.5 months.
Case 3
A woman of approximately 60 years of age underwent surgical resection of a pancreatic neck tumor in September 2019. The tumor measured approximately 2.4 × 1.3 cm, and microscopic and immunohistochemical analysis showed that it was a grade 2 (G2) p-NET with a Ki-67 index of 5%. No adjuvant therapy was administered following the surgery, and 2 years later, an abdominal MRI examination revealed the presence of multiple liver masses, with the largest measuring 2.1 cm, and multiple retroperitoneal lymph node masses. Subsequently, the patient underwent CAPTEM chemotherapy at our clinic. After completing three cycles of treatment in January 2022, she returned to the hospital for an evaluation of the treatment’s effectiveness. During the assessment, she complained of weakness in her lower limbs, which significantly affected her ability to walk steadily.
During the physical examination, the patient’s lower limb muscle strength was found to be poor, and she had hypertension (200/104 mmHg). Laboratory testing revealed severe hypokalemia (2.1 mmol/L), hyperglycemia (fasting blood glucose concentration of 7.31 mmol/L), high circulating ACTH and cortisol concentrations, with a lack of diurnal variation, and 24-hour urine cortisol production of >60 μg/day. However, she showed no classic signs of Cushing’s syndrome. Therefore, the patient underwent additional imaging examinations and liver biopsy. Abdominal enhanced MRI revealed the progression of the metastases in the liver and retroperitoneal lymph nodes, with the largest tumor measuring approximately 2.6 × 2.1 cm. Bilateral adrenal hyperplasia was also observed. Pituitary enhanced MRI did not indicate any abnormalities (Figure 2c, 2f, 2i). Examination of the liver biopsy revealed a grade 3 (G3) neuroendocrine tumor with a Ki-67 index of 22% and positive immunostaining for ACTH (Figure 3), implying the presence of EAS.

Histological images of the liver mass of case 3, stained with hematoxylin and eosin (HE) or immunohistochemical stains. (a) HE, original magnification ×400. (b, c) Tumor cells, immunostaining positive for synaptophysin and somatostatin receptor 2, original magnification ×400. (d) The Ki-67 index of tumor cells was approximately 22% and (e, f) Some tumor cells immunostained positive for adrenocorticotropic hormone, original magnification ×400.
In addition to symptomatic and anti-tumor therapy, mitotane (1,000 mg three times daily) was administered to manage the patient’s hypercortisolemia. After 14 days of treatment, her cortisol concentration had returned to normal (Figure 1); and her serum potassium, blood pressure, and blood sugar were under control; without any adverse events being recorded. Subsequently, the minimum necessary dose of mitotane for maintenance was determined through regular monitoring of the patient’s circulating cortisol concentration. Until the patient died on May 14, 2023, her hypercortisolemia remained under control through the use of mitotane (serum cortisol concentrations at 08:00 between 390 and 530 nmol/L). The patient survived for 44.6 months.
Discussion
The findings of the present case series suggest that mitotane alone can be used to control hypercortisolemia resulting from ectopic ACTH production in patients with p-NETs. This treatment showed promising results with respect to the symptoms and survival of the patients.
In the majority of patients, EAS is typically diagnosed synchronously, within a period of approximately 3 months before or after an initial diagnosis of a malignant tumor, whereas metachronous EAS, which is diagnosed more than 3 months after the initial diagnosis of a tumor, is relatively uncommon. However, some patients are diagnosed with EAS decades after Cushing’s syndrome is identified, when small tumors are found.7–9 It is noteworthy that patients with EAS do not always exhibit the clinical characteristics of Cushing’s syndrome. However, sudden severe hypokalemia is a key clinical feature of EAS, which is observed in approximately 42% to 86% of patients, and is a valuable diagnostic criterion. In the present study, two of the patients had synchronous EAS and one had metachronous EAS. All of them presented with advanced tumors, a substantial tumor burden, and severe hypokalemia. When diagnoses of p-NET-related EAS are made, the majority of patients have already developed distant metastases, with liver metastases being particularly common. 10 One possible explanation for this is that the ACTH secreted by pancreatic neoplasms is rapidly degraded in the liver after arriving via the enterohepatic circulation, and clinical symptoms only manifest when the tumors produce a sufficient quantity of hormone. 11 Another possible explanation is the heterogeneity of neuroendocrine tumors, the histopathological or endocrine properties of which can change when they metastasize, resulting in the tumor becoming the principal source of ACTH. This has been documented in several previous case reports.12,13 High circulating ACTH concentrations cause hypercortisolemia, which has a significant detrimental effect on the prognosis of patients and can be life-threatening. A previous systematic review of 336 patients with p-NET-related EAS that recorded the prognosis of 164 of these patients only followed them for 28.3 months (range 1 month to 20 years). 10 However, in this situation, the pharmaceutical control of hypercortisolism is mandatory.
Adrenal inhibitors of steroidogenesis, such as metyrapone, ketoconazole, etomidate, osilodrostat, and mitotane, are recommended by the Endocrine Society Clinical Practice Guidelines as first-line treatments for patients with metastatic EAS. 14 Of these, mitotane appears to be the most suitable option, on the basis of the present assessment. Ketoconazole use can be associated with hepatotoxicity, which poses a risk for patients with liver metastases; 15 metyrapone may mask biochemical hypoadrenalism and can occasionally cause hypertension, edema, and hypokalemia; 14 and etomidate requires continuous infusion and close monitoring, which is difficult to achieve. 16 A previous case report showed that abiraterone acetate can also significantly reduce cortisol concentrations, 17 but it can also aggravate the hypertension and hypokalemia of patients. Furthermore, it is unclear whether this drug can maintain cortisol concentrations of patients with unresectable advanced tumors within the normal range.
Mitotane is an analog of the insecticide dichlorodiphenyltrichloroethane and is the only medication approved by the United States Food and Drug Administration and the European Medicines Agency for the treatment of adrenocortical carcinoma. 18 It has a direct cytotoxic effect on the adrenal cortex through free radical-mediated metabolic transformation and oxidative damage, and inhibits adrenocortical steroid synthesis by inhibiting cholesterol side-chain cleavage and 11β-hydroxylase. 19 Mitotane also affects extra-adrenal cortisol bioavailability by increasing its hepatic clearance and the secretion of cortisol-binding protein, thereby reducing the availability of the hormone. 20 This complex mechanism of action makes it an effective treatment for hypercortisolemia, 21 and renders it a viable option for the management of hypercortisolism in such patients.
In a review by Donadille et al., data for 23 patients who underwent mitotane treatment at the Department of Endocrinology, Cochin Hospital were analyzed. Of these patients, four who had small-cell lung cancer did not undergo surgery and were followed for a median 1.025 years; 14 underwent surgical treatment and were followed for a median 11.66 years; and five individuals were found not to have tumors and were followed for a median 4.24 years. Only nine of these patients received mitotane as a single anti-cortisol medication. 22 Thus, there is currently limited experience with the use of mitotane as a standalone treatment for patients with advanced tumor-related EAS, and the available data do not conclusively demonstrate the benefits of this treatment for patients.
In the present study, three patients with advanced p-NET were diagnosed with EAS on the basis of a comprehensive assessment of their medical history, laboratory data, imaging findings, and response to anti-tumor therapy. Their excessive secretion of ACTH, resulting in adrenal gland hypertrophy and consequent hypercortisolism, was attributed to liver metastases that could not be completely eradicated. To treat this condition, in addition to administering anti-tumor therapy, we administered 3,000 mg/day mitotane to reduce the plasma cortisol concentrations of the patients and induce the destruction of their adrenal glands. Subsequently, we closely monitored the patients’ circulating hormone concentrations and those of other biochemical indices until they returned to normal, which happened after a mean of 9 days. Following long-term monitoring and extensive deliberation by our team, we gradually reduced the dose of mitotane to the minimum necessary to maintain the suppression of the patients’ hypercortisolism. Unfortunately, all three of the patients ultimately died because of tumor progression, but the median duration of follow-up was 39.6 months. It is worth noting that despite the ultimately negative outcomes, mitotane therapy was effective at maintaining the patients’ circulating cortisol concentrations within the normal range, despite tumor progression, and the duration of survival of the present patients was longer than that previously reported for patients with inoperable EAS.
The present study had a few limitations that should be addressed. First, the diagnostic process used could have been further strengthened to enhance the accuracy and reliability of the diagnoses made. Second, we did not measure the circulating concentrations of mitotane during the treatment period. Monitoring these concentrations could provide valuable information regarding the therapeutic efficacy of the drug and help optimize the dose administered.
Conclusions
This case series suggests that in situations where surgical intervention is not feasible, mitotane represents an effective and rapid means of controlling the life-threatening hypercortisolism associated with advanced p-NET. When used in combination with anti-tumor therapy, mitotane is well tolerated and has the potential to extend the survival of patients. However, further clinical trials are necessary to validate the effectiveness of mitotane treatment in such patients. Continued research in this area should provide valuable insights and contribute to the identification of the optimal treatment strategies for patients with advanced p-NET and ectopic ACTH syndrome.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605231220867 - Supplemental material for Effect of mitotane in patients with ectopic adrenocorticotropic hormone syndrome caused by advanced pancreatic neuroendocrine tumors: a case series and review of the literature
Supplemental material, sj-pdf-1-imr-10.1177_03000605231220867 for Effect of mitotane in patients with ectopic adrenocorticotropic hormone syndrome caused by advanced pancreatic neuroendocrine tumors: a case series and review of the literature by Shaobo Hu, Xianhua Wang, Fei Su, Qiong Zhou, Zhaoqing Li, Jie Luo and Huangying Tan in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605231220867 - Supplemental material for Effect of mitotane in patients with ectopic adrenocorticotropic hormone syndrome caused by advanced pancreatic neuroendocrine tumors: a case series and review of the literature
Supplemental material, sj-pdf-2-imr-10.1177_03000605231220867 for Effect of mitotane in patients with ectopic adrenocorticotropic hormone syndrome caused by advanced pancreatic neuroendocrine tumors: a case series and review of the literature by Shaobo Hu, Xianhua Wang, Fei Su, Qiong Zhou, Zhaoqing Li, Jie Luo and Huangying Tan in Journal of International Medical Research
Supplemental Material
sj-pdf-3-imr-10.1177_03000605231220867 - Supplemental material for Effect of mitotane in patients with ectopic adrenocorticotropic hormone syndrome caused by advanced pancreatic neuroendocrine tumors: a case series and review of the literature
Supplemental material, sj-pdf-3-imr-10.1177_03000605231220867 for Effect of mitotane in patients with ectopic adrenocorticotropic hormone syndrome caused by advanced pancreatic neuroendocrine tumors: a case series and review of the literature by Shaobo Hu, Xianhua Wang, Fei Su, Qiong Zhou, Zhaoqing Li, Jie Luo and Huangying Tan in Journal of International Medical Research
Footnotes
Acknowledgements
We thank the patients and their families for supporting the treatment of the patients.
Author contributions
SH performed the data analyses and wrote the manuscript; XW contributed to the manuscript preparation and the collection of clinical data; FS performed the review and revision of the manuscript; QZ contributed to the production of the figures and tables; ZL contributed to the analysis with constructive discussions; JL provided the pathological diagnosis and pictures; and HT contributed to the conception of the study and the analysis by constructive discussions.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Ethics statement
We obtained approval from the China-Japan Friendship Hospital ethics committee for the publication of this article. Because it was a retrospective study, the requirement for ethics approval was waived. All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee(s) and with the Helsinki Declaration (as revised in 2013). The authors are accountable for all aspects of the work, and will ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Written informed consent was obtained from the relatives of the patients for publication of this case series and the accompanying images.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.