
Abstract
Glycogen storage disease type 1b (GSD1b) is a rare genetic disorder, resulting from mutations in the SLC37A4 gene located on chromosome 11q23.3. Although the SLC37A4 gene has been identified as the pathogenic gene for GSD1b, the complete variant spectrum of this gene remains to be fully elucidated. In this study, we present three patients diagnosed with GSD1b through genetic testing. We detected five variants of the SLC37A4 gene in these three patients, with three of these mutations (p. L382Pfs*15, p. G117fs*28, and p. T312Sfs*13) being novel variants not previously reported in the literature. We also present a literature review and general overview of the currently reported SLC37A4 gene variants. Our study expands the mutation spectrum of SLC37A4, which may help enable genetic testing to facilitate prompt diagnosis, appropriate intervention, and genetic counseling for affected families.
Keywords
Introduction
Glycogen storage disease 1b (GSD1b OMIM 232220) is an autosomal recessive metabolic disorder distinguished by a deficiency of glucose 6-phosphate translocase (G6PT). The incidence rate of GSD1b is approximately 1 in 500,000 live births. 1 The SLC37A4 gene, which encodes G6PT, is found on chromosome 11 at 11q23.3. This gene spans 4.5 kb, comprises nine exons, and codes for a predominantly hydrophobic protein with 10 transmembrane domains. 2 G6PT plays a pivotal role in the translocation of glucose-6-phosphate (G6P) from the cytoplasm into the endoplasmic reticulum (ER) lumen. Subsequently, G6P undergoes hydrolysis to convert into glucose in the ER membrane. Additionally, G6PT expression is crucial for maintaining energy homeostasis in neutrophils. 3
The absence of G6PT activity leads to the buildup of glycogen and other metabolic intermediates in the liver, kidney, and intestines. This accumulation results in a variety of clinical manifestations. 4 The predominant clinical symptoms of GSD1b include hepatomegaly, hypoglycemia, lactic acidemia, hyperlipidemia, hyperuricemia, and growth retardation. Notably, G6PT is abundantly expressed in hematopoietic progenitor cells 5 and has a pivotal role in neutrophil homeostasis and its functional dynamics. 6 Defective G6PT can cause reduced glucose absorption that leads to a drop in the intracellular levels of G6P, ATP, lactate, and NADPH. 3 Thus, GSD1b in G6PT brings about neutropenia and neutrophil dysfunction and a higher susceptibility to various infectious diseases, including recurrent bacterial infections and inflammatory bowel disease (IBD). 7
Since the SLC37A4 gene was first identified, more than 100 variants have been reported, with the missense mutation being the most common variant. 8 Further analysis of the SLC37A4 gene may provide an important clinical strategy for patients with GSD1b. Despite the identification of SLC37A4 as the pathogenic gene for GSD1b, a comprehensive understanding of the complete spectrum of its genetic variants remains to be fully elucidated. In this study, we have identified three novel SLC37A4 variants. We then systematically review the literature pertaining to reported variants to further clarify the full variant spectrum of SLC37A4.
Case presentations
Patient 1
Patient 1 (male) was admitted to the hospital at the age of 17 months from the presence of abdominal distension. He had a medical history characterized by recurrent episodes of pneumonia, bronchitis, and hypoglycemic attacks since the age of 3 months. Additionally, he experienced chronic diarrhea from birth until the age of 1 year. Upon admission, the patient's height measured 69 cm, corresponding to a height-for-age z-score of −4.9 (0.1st percentile). His weight was 9 kg, resulting in a weight-for-age z-score of −1.8 (4th percentile). A physical analysis revealed the presence of hepatomegaly. Notably, the fasting glucose, lactic acid, and blood triglyceride (TG) levels were found to be abnormal (measuring 1.6 mmol/L, 12.35 mmol/L, and 12.35 mmol/L, respectively). Moreover, urine ketone bodies tested positive. An abdominal ultrasound analysis confirmed the presence of hepatosplenomegaly. Subsequent genetic analysis identified a compound heterozygous variant on the SLC37A4 gene, consisting of a c.1145_1163del
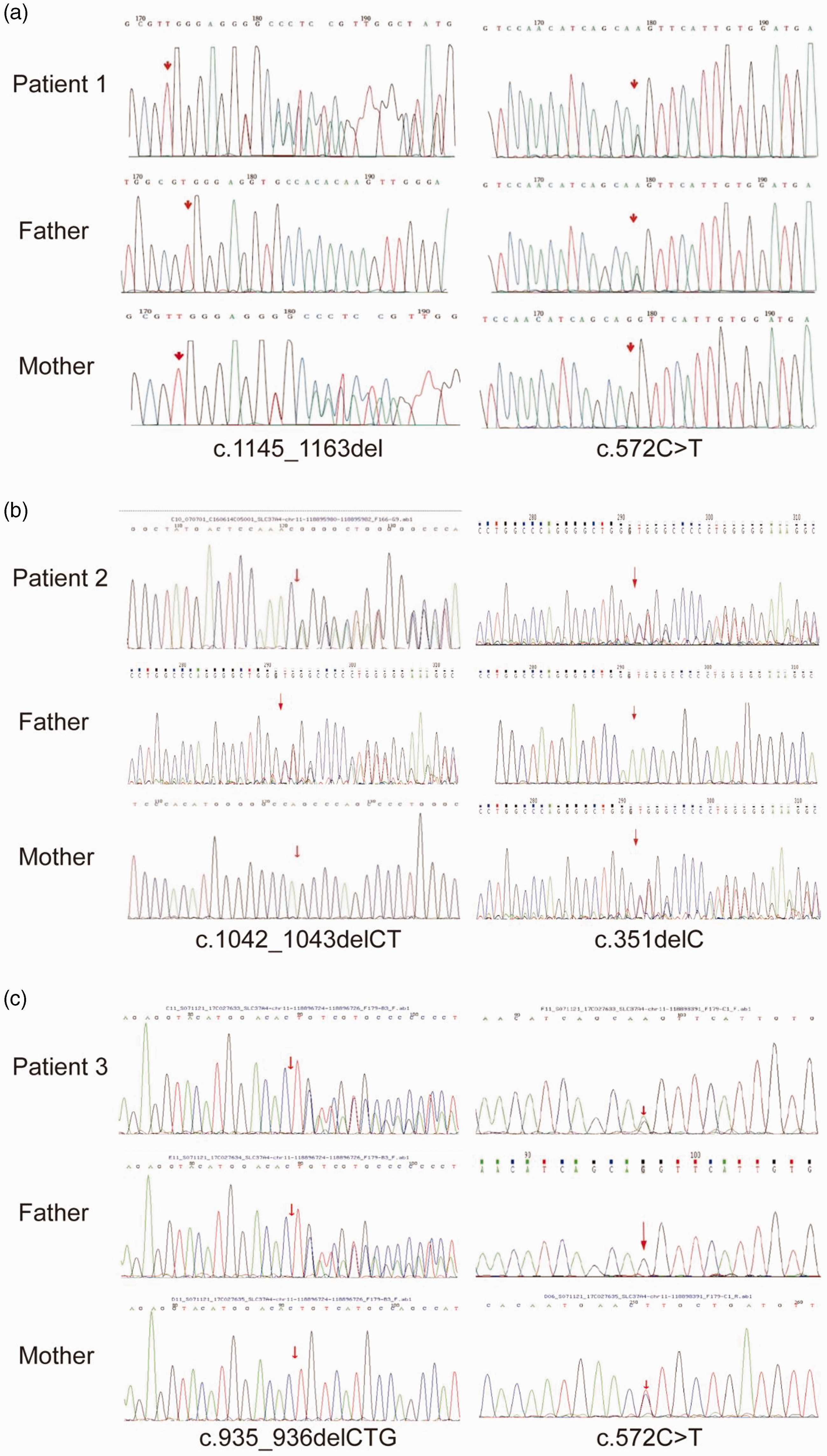
Sanger sequencing results and sequence conservation analysis. (a) The novel variant c.1145_1163del

Endoscopy images of Patient 1 (a, b) and Patient 2 (c, d). (a) An ileocecal ulcer in the ileocecal area. (b) Hyperemia and edema of the ascending colon. (c) A large ulcer in the ileocecal area and (d) Mucosal erosion in the transverse colon
Patient 2
At the age of 2 months, a male patient was admitted to the hospital from pneumonia and poor spirit. During his hospitalization, the patient presented with a hypoglycemic state, with low blood glucose levels of 2.0 mmol/L and high blood lactic acid levels of 7.7 mmol/L. Considering the clinical symptoms, it was suspected that the patient harbored inborn errors of metabolism. We then performed genetic testing, which confirmed that the patient had a compound heterozygous variation of c.1042_1043delCT (p.L348Vfs*53) in exon 10 and c.351delC (p. G117fs*28) in exon 4 of the SLC37A4 gene. The former variation was inherited from the father, while the latter was inherited from the mother (Figure 1b). The c.351delC variant was a previously unreported mutation. Using the ACMG criteria, the variant was categorized as “likely pathogenic” (PVS1+ PM2_supporting). Sanger sequencing revealed that the mother carried this variant in a heterozygous state at this site, while the father did not exhibit any variant at this site (Figure 1b). The SIFT and PolyPhen-2 prediction programs indicated that the variant was likely harmful, with p.G117fs*28 representing a frameshift variant that could potentially impact the structure and function of the SLC37A4 protein. To manage this condition, the patient was administered lactose-free milk powder and his feeding frequency was augmented. At the age of 16 months, he commenced oral intake of uncooked cornstarch at a dosage of 2 g/kg/day every 6 hours. However, despite these interventions, his blood glucose levels remained unstable, plummeting to 3.1 mmol/L. Additionally, the patient experienced recurrent oral ulcers and gingivitis approximately three to five times per year, starting at the age of 2.5 years. These oral lesions persisted for a month and exhibited a protracted healing process. The patient also suffered from respiratory tract infections three to five times per year, which exhibited improvement upon antibiotic therapy. At the age of 3 years and 5 months, the patient presented with mucopurulent bloody stool and an increased frequency of defecation. During his subsequent hospitalization, laboratory test results revealed normal liver function but an abnormal blood routine (WBC 5.2 × 109/L, NE 0.61 × 109/L, platelet (PLT) 438 × 109/L). However, colonoscopy unveiled edema of the ileocecal valve and appendicular ostium, accompanied by mucosal erosion and a sizable ulceration, as well as erosions in the transverse colon (Figure 2c, d).
After G-CSF treatment, the patient's WBC count increased and the ANC returned to normal. Two months later, his appetite improved, the daily stool frequency reduced to once per day, and the mucopurulent bloody stool disappeared. His weight increased by 3 kg. The medication was self-discontinued. At 2 years and 6 months old, abnormal bowel movements occurred six to seven times daily, without blood or mucus. Recurrent oral aphthous ulcers with slow healing were also present.
At the age of 6 years, the patient presented with recurring episodes of vomiting, chronic diarrhea, and oral ulcerations. In addition, he displayed an inability to maintain neutrophil levels in the normal range. Therefore, the patient was prescribed empagliflozin at a dosage of 10 mg/day (0.4 mg/kg/day) and administered uncooked cornstarch at a dosage of 48 mg (2 mg/kg/day) every 4 hours. Following a 2-month course of treatment, the neutrophil levels gradually trended towards normalcy. Concurrently, his symptoms of diarrhea and vomiting significantly abated and the occurrence of recurrent oral ulcerations ceased. The patient's height measured 115 cm, corresponding to a height-for-age z-score of −1.17. Additionally, the patient's weight was recorded as 24 kg, yielding a weight-for-age z-score of 0.32.
Patient 3
A 5-month-old boy was admitted to the hospital with pneumonia and diarrhea. The patient had experienced diarrhea since the age of 2 months, with six to seven loose stool episodes per day, but received no medication. Prior to admission, the patient had a fever and cough for 10 days. He had a height of 63 cm and weight of 7.5 kg, with a weight-for-length z-score of 1.2 (88th percentile). The patient presented with a fever exceeding 39°C, tachycardia of 200 beats per minute, tachypnea of 64 breaths per minute, bilateral wheezing and rhonchi in the lungs, hepatomegaly measuring 8 cm below the right costal margin, and splenomegaly measuring 2 cm below the left costal margin.
The blood tests revealed a base excess of −20.4 and a cHCO3- level of 0.5 mmol/L. His fasting blood glucose measured 2.52 mmol/L, lactate was recorded at 5.33 mmol/L, and urine ketone test yielded a positive result. The AST and ALT levels were elevated, with values of 266 U/L and 240 U/L, respectively. Pneumonia-related indications were observed in the chest X-ray. Throughout the hospitalization period, the lowest recorded blood glucose level was 1.8 mmol/L. Sanger sequencing of all the exons and exon-intron boundaries of the SLC37A4 gene (NM_001164277) in the proband detected a compound heterozygous variant, including c.935_936delCTG (p.T312Sfs*13) in exon 9 inherited from the father and c.572C>T (p.Pro191Leu) in exon 6 inherited from the mother. The c.935_936delCTG has not previously been reported. Using the ACMG guidelines, the present variation was initially classified as a suspected pathogenic variant (PVS1+PM2_Supporting). Sanger sequencing revealed that the father was heterozygous for this site, while the mother did not possess any variant at this site (Figure 1c). The SIFT and PolyPhen-2 prediction programs indicated that the variant was likely harmful, with the p.T312sfs*13 mutation representing a frameshift mutation that may affect the structure and function of the SLC37A4 protein. Subsequent treatment for the patient involved the administration of antibiotics and oral uncooked cornstarch. This intervention led to an improvement in clinical symptoms, thus resulting in the patient's discharge.
Table 1 presents the analysis results of all three patients during their initial hospitalization. Verbal informed consent and treatment consent were obtained from the parents of all patients. The reporting of this study adheres to the CARE guidelines. 9
Examination results of the patients at first hospitalization.

WBC, white blood cell; ANC, absolute neutrophil count; PLT, platelet; AST, aspartate aminotransferase; ALT, alanine aminotransferase; TG, triglyceride; TC, total cholesterol.
Normal range: WBC, 4.0–10.0 × 109/L; ANC > 1.5 × 109/L; PLT, 150–400 × 109/L; ALT, 4–41 U/L; AST, 4–40 U/L; TG, 0.05–1.7 mmol/L; TC, 2.9–5.68 mmol/L; fasting glucose, 3.61–6.61 mmol/L; lactic acid, 0.50–2.20 mmol/L; uric acid, 202.3–416.5 μmol/L.
Literature review
A literature review was conducted to retrieve all previously published SLC37A4 variants in GSD1b from 1978 to 2023 from the MEDLINE, PubMed, and Web of Science databases. Upon reviewing the literature, a total of 134 mutations were identified for the SLC37A4 gene (Table 2). Among these variants, the most prevalent one was the c.1042_1043delCT (p. Leu348Vfs*53) mutation (Table 2). Additionally, 25 variants have been reported in the Chinese population, with the c.572C>T (p. Pro191Leu) variant being the most frequent. The overall spectrum of SLC37A4 variants is summarized in Table 2, which includes references not cited in the main text.
The spectrum of SLC37A4 variants
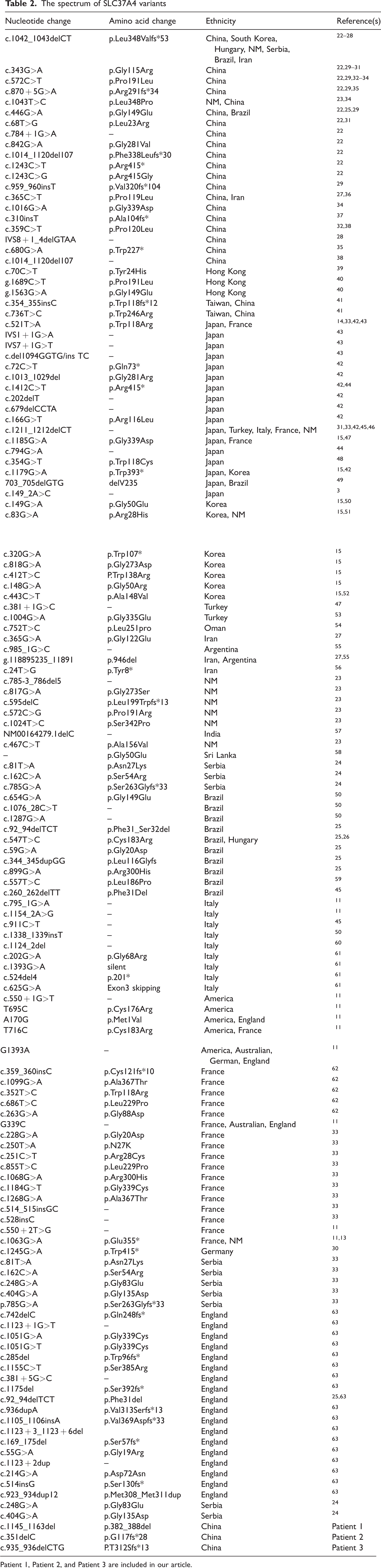
Patient 1, Patient 2, and Patient 3 are included in our article.
Discussion and conclusions
GSD1 comprises a group of rare hereditary disorders resulting from an insufficiency in the glucose-6-phosphatase (G6Pase) system. This system plays a crucial role in maintaining glucose homeostasis by facilitating the hydrolysis of G6P into glucose and inorganic phosphate (Pi). 10 Of the two types of GSD1, glycogen storage disease type 1a (GSD1a) is the most prevalent, accounting for approximately 80% of GSD1 patients. It is caused by variants in the G6Pase gene, while the remaining 20% of GSD1 cases are diagnosed as GSD1b. 11
GSD1b arises from a deficiency in G6PT. 12 The SLC37A4 gene encodes G6PT, which facilitates the transport of G6P from the cytoplasm to the ER lumen and subsequently delivers it to the catalytic site of G6Pase. Situated on human chromosome 11q23.3, the SLC37A4 gene comprises nine exons, spans approximately 5.3 kb of genomic DNA, and exhibits high expression in the liver, kidney, intestines, and skeletal muscle. Firstly, Gerin et al. identified the p.W118R variant in the putative G6P transporter in two patients, 13 a finding later corroborated by Kure et al. 14 To date, a total of 132 pathogenic variants have been characterized in the SLC37A4 gene, all of which are associated with GSD1b. The most prevalent variant, c.1042_1043delCT (p. Leu348Valfs*53), has been frequently observed in mixed Caucasian (27% to 31%) and German (32%) populations. Consistent with previous reports, Patient 1 in our study also had this variant. The literature has indicated that the type and proportion of variants in GSD1b may vary across different ethnic groups. In the Korean population, the most common variant is c.443C > T (p. Ala148Val), present in 55.6% of GSD1b patients and 38.9% of alleles. 15 In the Japanese population, the predominant variant is c.352 T > C (p. Trp118Arg), detected in 37% to 50% of GSD1b patients. 7 Among the Chinese population, 25 variants have been identified, with c.572C>T (p. Pro191Leu) being the most frequent, accounting for 18.8% of alleles. Notably, this variant has not been reported in other ethnic groups with GSD1b, suggesting its potential significance as a diagnostic marker specifically for GSD1b in the Chinese population.
The clinical features of GSD1 include hypoglycemia, growth retardation, hyperlipidemia, and lactic acidemia. 7 Additionally, individuals with GSD1b may exhibit neutropenia, neutrophil dysfunction, recurrent bacterial infections, and IBD. 15 According to reports from various geographical regions, neutropenia affects over 94% of GSD1b patients. 3 In these cases, neutropenia was observed in all three patients. Among GSD1b patients, IBD is regarded as one of the most severe complications. The incidence of IBD is on the rise, with the mean age at initial diagnosis being 8.7 years. 16 Nearly all patients experienced abdominal pain or symptoms indicative of intestinal obstruction, with 60% of them presenting with ileal or colonic strictures, necessitating surgical resection or endoscopic dilatation in 50% of cases. 16 Research by Mikami et al. revealed that 28% of patients had confirmed IBD, while an additional 22% exhibited symptoms highly suggestive of IBD, which significantly impacted their quality of life and overall well-being. 17 Many GSD1b patients with IBD manifest chronic gastrointestinal inflammation, characterized by recurrent abdominal pain, emesis, frequent or persistent diarrhea, or the presence of draining fistulas. In our cases, all three patients presented with gastrointestinal symptoms, and two of them were diagnosed with IBD. At the age of ten, Patient 1 experienced recurrent diarrhea and abdominal pain 8 years after his GSD1b diagnosis. Patient 2, at 3 years and 5 months old, exhibited mucopurulent bloody stool with an increased frequency of defecation three years after his GSD1b diagnosis. Patient 3 sought medical attention at our hospital following a 3-month history of diarrhea. Colonoscopy and pathological analysis confirmed the diagnosis of IBD in Patient 1 and Patient 2. The underlying cause of IBD in GSD1b patients may be attributed to neutrophil dysfunction and reduced neutrophil count. The absence of neutrophils in the intestinal mucosa renders the gastrointestinal tract susceptible to pathogenic and commensal bacteria, thus leading to IBD development. 18
For therapy, the metabolic abnormalities in GSD1b can be managed through dietary interventions aimed at maintaining normoglycemia and alleviating symptoms. However, these interventions have limited efficacy in preventing long-term complications such as renal disease and hepatocellular adenoma/carcinoma. Traditional treatment involving subcutaneous injections of G-CSF has been shown to enhance neutrophil numbers but not their functions, and may even elevate the risk of developing monoclonal malignancies, including myelodysplasia and acute myeloid leukemia. 19 Recent studies have indicated that empagliflozin, a sodium glucose co-transporter 2 inhibitor, represents a safe and promising alternative for treating neutropenia in individuals with GSD1b, IBD, and neutropenia. 20 In this study, empagliflozin was prescribed to Patient 2 at the age of 6 years, leading to a gradual discontinuation of G-CSF. Subsequently, the patient experienced an improvement in gastrointestinal symptoms, with a normal ANC observed during the last follow-up and no reported adverse effects. Notably, no severe adverse events (AE) were documented throughout the treatment period. A retrospective analysis involving 112 patients from 24 countries demonstrated the efficacy of empagliflozin in ameliorating neutropenia and its favorable outcomes in relation to various symptoms associated with neutropenia and neutrophil dysfunction. These symptoms encompassed oral and urogenital mucosal lesions, recurrent infections, skin abscesses, IBD, and anemia. Among the participants, hypoglycemia was the most commonly reported AE, with a prevalence of 18%. 21 In addition, glycolysis serves as the sole energy source for mature neutrophils. The precursor of toxic 1,5-AG6P, which is the structural analog of G6P, is 1,5-anhydroglucitol (1,5-AG). Its dephosphorylation is facilitated by G6PT. Because of the deficiency of G6PT, the initial step of glycolysis is inhibited, resulting in 1,5-AG6P accumulation in the cytoplasm. 6 Wortmann et al. observed a four- to five-fold decrease in plasma 1,5-AG levels following daily empagliflozin intake in all patients. This was subsequently followed by the establishment of a new stable 1,5-AG concentration after 2 to 3 weeks. Prior to treatment, only 34% of neutrophils exhibited an oxidative burst response similar to that of the healthy control sample. Surprisingly, after treatment, 96% of neutrophils displayed oxidative bursts, accompanied by a subsidence of IBD symptoms, thereby enabling the gradual discontinuation of G-CSF treatment. Therefore, neutrophil counts were significantly elevated and gradually approached the normal range. 18
In this study, three novel SLC37A4 genetic variants in GSD1b patients were reported, expanding the variant spectrum of this gene and enhancing our understanding of the disease. Genetic analyses may facilitate the diagnostic process for GSD1b, potentially leading to early diagnosis and therapeutic intervention that could possibly improve patient quality of life.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605231216633 - Supplemental material for Three novel SLC37A4 variants in glycogen storage disease type 1b and a literature review
Supplemental material, sj-pdf-1-imr-10.1177_03000605231216633 for Three novel SLC37A4 variants in glycogen storage disease type 1b and a literature review by Zhuolin Wang, Ruiqin Zhao, Xiaoyun Jia, Xiaolei Li, Li Ma and Haiyan Fu in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605231216633 - Supplemental material for Three novel SLC37A4 variants in glycogen storage disease type 1b and a literature review
Supplemental material, sj-pdf-2-imr-10.1177_03000605231216633 for Three novel SLC37A4 variants in glycogen storage disease type 1b and a literature review by Zhuolin Wang, Ruiqin Zhao, Xiaoyun Jia, Xiaolei Li, Li Ma and Haiyan Fu in Journal of International Medical Research
Footnotes
Acknowledgements
We appreciate the help of all our colleagues in our hospital.
Author contributions
ZW analyzed the data and wrote the first draft of the manuscript. HF designed the research. XJ, XL, and LM analyzed the genetic testing results. RZ edited and reviewed the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Data availability statement
Data are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
All authors declare that there is no conflict of interest.
Ethics approval and consent to participate
The study was approved by the Ethical Committee of the Children’s hospital of Hebei Province in China (Approval No. 2021170). We also obtained informed consent from all patients’ parents.
Funding
This work was supported by the Hebei Province Medical Science Research Project (No. 20210246) and Hebei Provincial Government-Funded Clinical Medicine Talent Training Project (No. ZF2024186).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.