
Abstract
Despite the widespread use of early revascularization and drugs to regulate the neuroendocrine system, the impact of such measures on alleviating the development of heart failure (HF) after myocardial infarction (MI) remains limited. Therefore, it is important to discuss the development of new therapeutic strategies to prevent or reverse HF after MI. This requires a better understanding of the potential mechanisms involved. HF after MI is the result of complex pathophysiological processes, with adverse ventricular remodeling playing a major role. Adverse ventricular remodeling refers to the heart’s adaptation in terms of changes in ventricular size, shape, and function under the influence of various regulatory factors, including the mechanical, neurohormonal, and cardiac inflammatory immune environments; ischemia/reperfusion injury; energy metabolism; and genetic correlation factors. Additionally, unique right ventricular dysfunction can occur secondary to ischemic shock in the surviving myocardium. HF after MI may also be influenced by other factors. This review summarizes the main pathophysiological mechanisms of HF after MI and highlights sex-related differences in the prognosis of patients with acute MI. These findings provide new insights for guiding the development of targeted treatments to delay the progression of HF after MI and offering incremental benefits to existing therapies.
Keywords
Introduction
Heart failure (HF) is usually the end stage of cardiovascular disease. Its prevalence is increasing, which is largely influenced by the increased survival rate of patients with myocardial infarction (MI).1,2 For example, risk factors such as diabetes, chronic obstructive pulmonary disease, hypertension, and chronic kidney disease can accelerate the progression of HF. Patients with these risk factors can develop cardiac insufficiency or HF after MI, which can increase the risk of death or adverse outcomes including cardiac rupture, cardiac arrest, ventricular arrhythmias, recurrent MI, and even sudden death.3,4 These adverse outcomes increase the economic pressure on the healthcare system and hinder effective medical management. Therefore, it is essential to understand the pathogenesis of HF after MI to improve its diagnosis and treatment, prolong patients’ lifespan, and decrease the risk of adverse outcomes. 5
The myocardium is composed of cardiomyocytes (CMs), capillaries, and extracellular matrix (ECM), all of which help to maintain normal cardiovascular physiological function. HF after MI is caused by structural and functional abnormalities of the heart that occur following acute MI (AMI) (including ST-segment elevation MI and non-ST-segment elevation MI) during hospitalization or after discharge. The pathological changes in cardiac structure and function are commonly referred to as adverse ventricular remodeling (AVR). This process involves complex and progressive molecular and cellular transformations that markedly increase the risk of HF and reduce the survival rate. 6
In this review, we summarize the main pathophysiological mechanisms of AVR after MI and examine the currently available diagnostic methods and treatments, particularly highlighting the sex-related differences in the prognosis of patients with AMI. We also discuss the direction of research in this field in the context of clinical applications.
AVR
In most cases, the development of HF after MI originates from AVR of the left ventricle (LV). This is likely explained by much higher stroke work of the LV than right ventricle (RV) because of the former’s dominance in cardiac pumping function. AVR after MI is characterized by the heart’s adaptations in terms of ventricular size, shape, and function in response to comprehensive regulatory factors such as the mechanical, neurohormonal, and cardiac inflammatory immune environments; ischemia/reperfusion (I/R) injury; energy metabolism; and genetic correlation factors.7–9 The structure and function of CMs, capillaries, and ECM are maladaptive under the above regulatory mechanisms, ultimately leading to increased cardiac stiffness and deterioration of diastolic and systolic function (Figure 1).

Mechanism of cardiac remodeling.
Mechanical agents
The geometric changes of the ventricle are the main stimulus for LV remodeling. Laplace’s law states that ventricular wall stress is directly related to the LV pressure and radius and inversely proportional to twice the LV wall thickness. In the early stage of MI, the cardiac load increases as a compensatory adaptive response to mechanical and physiological stress that damages cardiac output, causing CM hypertrophy and subsequent myocardial hypertrophy. Loss of cardiac systolic function and a secondary increase in LV volume can increase wall stress and oxygen consumption, which eventually results in maladaptive tissue remodeling and triggers the transition to HF. 10 This transition to HF is triggered by attempts to compensate for the increased preload and afterload. The passive mechanical restraint of the surrounding normal myocardium also mediates a decrease in the systolic function of the infarcted area and even the overall ventricle. 11
Neurohormonal system regulation
The integrated regulation by the neurohormonal system plays an important role in maintaining the physiological balance of the cardiovascular system and is a critical link in the development of AVR leading to LV dysfunction. The cascade of cardiac regulatory hormones related to LV remodeling is comprehensively regulated by the sympathetic nervous system (SNS) and the renin–angiotensin–aldosterone system (RAAS), and the natriuretic peptide system provides a beneficial compensatory effect. 8
Reduced cardiac output after MI due to acute decompensation can reflexively activate the SNS and provide β-adrenergic tone, resulting in compensatory enhancement of myocardial contractility, an increased heart rate, and increased myocardial oxygen consumption. 12 However, sustained sympathetic activation is detrimental to the LV, mainly because of the toxic effects of high concentrations of its downstream products (catecholamines such as norepinephrine and epinephrine) on CMs, as well as stimulation of fibroblast synthesis to promote myocardial interstitial fibrosis; these processes eventually lead to the development of HF.13–18 Chronic SNS activation is a key promoter of RAAS activation, and reduced cardiac output also reflexively activates the RAAS, thereby mediating the adverse effects of angiotensin II. The binding of angiotensin II to angiotensin II type 1 receptors in the circulation causes increased aldosterone secretion, promoting AVR as well as leading to cell apoptosis and interstitial fibrosis, thus accelerating the development of HF. 15 As aldosterone decreases the sensitivity of pressure receptors, the inhibition of sympathetic activity is reduced and norepinephrine release is increased. This suggests that attenuation of the pressure reflex by local aldosterone release may trigger sympathetic activation. 19 In addition, the decrease in cardiac output activates the natriuretic peptide system to exert a beneficial compensatory effect. The three identified natriuretic peptide hormone subtypes are type A, B, and C natriuretic peptide. They can inhibit the SNS and RAAS to exert a beneficial compensatory effect.20,21 These natriuretic peptides can dilate vessels while reducing pulmonary capillary wedge pressure, systemic arterial pressure, and RV pressure, and the resultant decrease in cardiac preload and afterload can improve cardiac remodeling.
The level of SNS and RAAS activation is correlated with the severity, outcome, and prognosis of HF. Thus, angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers, β-blockers, mineralocorticoid receptor antagonists, and other drugs have been widely used in clinical practice to improve AVR. ACEI therapy has shown survival benefits after MI. Early initiation of ACEI therapy after MI improves short-term mortality, and administration of captopril after AMI can reduce the LV end-diastolic volume (LVEDV) and LV filling pressure.22–24 The improvement in clinical outcomes using angiotensin receptor blockers and ACEIs is similar.25,26 Use of β-blockers has consistently been shown to provide a survival advantage in patients with HF, and it plays a crucial role in β-adrenergic blockade in infarct remodeling.27–29 Moreover, mineralocorticoid receptor antagonists are considered central molecules in ECM regulation, especially during collagen deposition. 30 Moreover, natriuretic peptides and nitric oxide (NO) can synchronously activate cyclic GMP-dependent protein kinases to exert anti-hypertrophic effects, and clinical studies of the cyclic GMP-phosphodiesterase inhibitor sildenafil, which can stimulate NO production, have shown its beneficial effects in the treatment of congestive HF.31–33
Inflammatory immune activation
AMI triggers the inflammatory response, which aims to remove necrotic cellular debris and initiate anti-inflammatory repair. Cellular necrosis and apoptosis are essential for inflammatory immune system activation. Although these processes occur simultaneously, apoptosis is programmed cell death without the release of intracellular components whereas necrosis is an uncontrolled form of cell death with cell rupture. Later, the intracellular components released by the necrotic cells activate the immune system through innate immune receptors, resulting in infiltration of inflammatory cells. The differentiation and interaction of the various inflammatory cell subtypes are strictly regulated. By mediating inflammation or healing, inflammatory immune system activation plays a dual role: the chronic inflammatory state drives maladaptive remodeling, but infiltrating inflammatory cells can recruit leukocytes, remove tissue debris, and start the remedial response, thus allowing scar tissue formation. 34 However, inflammation can also mediate the release of damage-associated molecular patterns (DAMPS) from necrotic and apoptotic CMs after MI, continuously activating the innate immune system and triggering more severe and overactive inflammatory reactions. 35 The binding of DAMPs to pattern recognition receptors or Toll-like receptors is crucial for the activation of proinflammatory signaling pathways. Persistent dysregulated inflammation can promote CM death, impair the contractile function of surviving CMs, and promote interstitial tissue destruction. 36 Studies have shown that the inflammatory response is directly proportional to the degree of myocardial injury as evidenced by elevated levels of systemic inflammatory markers, such as C-reactive protein, being associated with a worse prognosis in patients with AMI and HF.37–39
Different inflammatory cells and large numbers of cytokines and matrix metalloproteinases (MMPs) facilitate molecular coordination of the myocardial inflammatory response through extensive interactions, leading to expansion of the range of damage, aggravation of impaired tissue function, and acceleration of HF. Among these interactions, neutrophils and macrophages are major players in remodeling and are the primary leukocytes that infiltrate the infarcted LV. 40 Monocytes infiltrate first, after which neutrophils rapidly become the predominant leukocytes in the infarcted area. Although neutrophil infiltration reduces injury after AMI, it exacerbates the fibrotic response, promoting progression to HF.41,42 Macrophages act as phagocytes to remove necrotic debris and apoptotic neutrophils, and their depletion can exacerbate LV remodeling and over-expansion. 43 The absence of polarization components of macrophages, such as CCR5 deficiency, does not limit the infiltration of these cells into the infarcted area; however, it hinders the healing process and increases the risk of rupture induced by proinflammatory and anti-inflammatory proteins. 44 Lymphocytes and mast cells also play an important role in the activation of fibroblasts and local inflammation within the myocardium.45–48 Endothelial cells can also promote the formation of a pro-fibrotic inflammatory environment by activating a pro-inflammatory secretion phenotype. 49
Current studies are focusing on the use of different cytokines and chemokines as potential targets to improve cardiac function and the prognosis in patients with AMI or HF, such as inhibition of interleukins 1, 6, and 8; monocyte chemotactic protein 1; CC and CXC chemokines; and tumor necrosis factor α. 50 Because elevated MMPs after MI regulate remodeling by promoting ECM conversion and inflammatory signaling and are closely related to LV dysfunction in patients with HF, they may also serve as biomarkers of MI or HF and as future targets for preventing HF after MI. 51
I/R injury, reactive oxygen species (ROS), and mitochondrial energy metabolism
Although emergency coronary intervention is beneficial for patients with AMI, reperfusion to restore myocardial blood flow allows the infarct size to expand, causing a series of harmful metabolic changes in CMs. 52 This process is referred to as I/R injury and is characterized by a series of continuous and reciprocal processes. At the molecular level, reperfusion leads to sudden oxidization of succinate that accumulates during ischemia, mediating the production of ROS. The sustained increase in the oxygen free radical concentration can create a vicious circle through MMP activation followed by chronic mitochondrial remodeling, which reduces energy production and ultimately promotes the development of HF.53,54
After MI, CMs are exposed to acute ischemic and hypoxic environments. Because anaerobic glycolysis becomes the only important source of newly generated adenosine triphosphate during coronary artery occlusion, a large amount of H+ can be generated simultaneously and accumulate intracellularly. Once perfusion is restored, H+ is transported to the extracellular space to ensure normal pH; this results in an increase in the intracellular Na+ concentration, which can eventually lead to Ca2+ overload. 55 High calcium concentrations induce opening of the mitochondrial permeability transition pore (mPTP), while the opening of nonselective channels in the inner mitochondrial membrane is a crucial determinant of lethal I/R injury. These channels open only in the first few minutes after myocardial reperfusion in response to mitochondrial Ca2+ overload, oxidative stress, physiological pH recovery, and adenosine triphosphate depletion. The formation and opening of mPTP during myocardial I/R can lead to outer membrane rupture because of the inner membrane potential depolarization and matrix expansion. Eventually, substances such as proteins and cytochrome C can be released into the cytoplasm to activate the cysteine cascade reaction, initiating cell death. 56 The accumulation of intracellular H+ and Ca2+ and destruction of the mitochondrial membrane potential can also lead to significant oxygen-derived free radical production during I/R injury. The chronic accumulation of oxyradicals in mitochondria can lead to a catastrophic cycle of mitochondrial DNA damage and decreased mitochondrial function, further generating ROS. These ROS can induce CM hypertrophy, CM apoptosis, and interstitial fibrosis through the activation of MMPs, forming a vicious circle of AVR and progression of HF.7,54,57 MMP and tissue inhibitor of metalloproteinase are coordinately regulated at the transcriptional and translational levels through different transcription factors and enzymes, including the NF-κB and JAK-STAT pathways, and their regulation can also be influenced by neurohormonal activation. 58 High circulating levels of MMPs are associated with dilative AVR, systolic dysfunction, and an increased risk of progression to HF.59,60 Progressive metabolic remodeling is a key driver of the transition to HF after MI, which will exacerbate the occurrence and development of AVR. 61
Genetic regulation
In addition to the numerous above-described proven pathophysiological mechanisms leading to the development of HF after MI, genetically related regulatory mechanisms have been continuously revealed. Among these, epigenetic regulatory mechanisms play a major role.
Decreased CM contractility has been found to involve an altered cell phenotype. Pressure overload leads to a disproportionate increase in the ventricular wall thickness, and the expression of genes such as myosin and troponin can show embryonic alterations. 62 This is widely related to the abnormal expression of certain microRNAs. For example, microRNA-208 encoded by the α-major histocompatibility complex gene plays a significant regulatory role in the production of myosin heavy chains, and its upregulation can lead to CM hypertrophy through negative regulation of SOX6. 63 Epigenetic regulation of non-coding RNAs, including long-stranded non-coding RNAs and microRNAs, play an essential role in I/R injury through endoplasmic reticulum (ER)–mitochondria microdomain interactions. 64 The sensitivity of CMs to death is fine-tuned by the intracellular calcium concentration. Therefore, when inositol 1,4,5-trisphosphate receptor (IP3R)-mediated I/R stress upregulates and leads to mitochondrial calcium overload, necrotic apoptosis signal pathways will be activated through the CaMKII-mPTP or XO-ROS-mPTP pathway in the reperfused heart. There is also evidence to suggest that receptor-interacting protein kinase 3 (Ripk3) manages IP3R located in the ER. Because genetic ablation of Ripk3 eliminates reperfusion-induced IP3R upregulation and ER stress confirms the necessity of ER–mitochondrial microdomains in stimulating Ripk3-induced necrotic apoptosis in cardiac I/R injury. 65 Additionally, the downstream event of necroptosis is the opening of Ripk3-activated mPTP, which mediates the swelling and rupture of organelles and cells due to energy generation obstacles. Interestingly, inhibition of certain genes also restores the adult heart to the metabolic state of the fetal gene pattern. 66
Multiple studies have focused on the susceptibility of individuals to I/R injury, which may be related to genes encoding angiotensin II, ACE, osteoprotegerin, transforming growth factor β, vascular endothelial growth factor, galactose lectin-3, ficolin-1, mitochondrial aldehyde dehydrogenase 2, and others. 67
Cardiac adaptive changes under integrated regulation
Cardiac adaptive changes mainly involve changes in ventricular size, shape, and function, which are primarily caused by CM changes and myocardial fibrosis in the infarct area, infarcted marginal area, and non-infarcted area. Physiological hypertrophy of CMs is often described as volume expansion of terminally differentiated CMs, a process that can resolve without causing any permanent damage. By contrast, the triggering of a series of pro-hypertrophic molecular pathways by MI often leads to overt myocardial damage. CMs may exhibit pathophysiological changes such as hypertrophy, necrosis, and apoptosis after MI. In the early stage of injury, the collagen scaffold is disrupted because inflammatory cells flow into the necrotic myocardium, making it challenging to maintain the ventricular shape and causing localized myocardial thinning and dilation in the infarcted region.68,69 Because of the increased volume and pressure load, the surviving CMs in the non-infarcted region undergo stretching and compensatory eccentric hypertrophy, 70 which further leads to LV dilation. Compared with distal CMs, those in the infarcted marginal area are located closer to the hypertrophic infarcted myocardium and exhibit more severe differentiation, sarcomere depletion, and reduced mitochondrial volume. Additionally, myocardial fiber stretching and inflammation are essential triggers for myofibroblast activation. Necrotizing apoptosis of CMs after MI stimulates the development of myofibroblasts and results in collagen deposition, leading to progressive alternative fibrosis and eventually forming scars mainly composed of cross-linked collagen; these are considered irreversible changes. 71 In the non-infarcted region, increased wall stress can also locally activate CMs, macrophages, and fibroblasts, causing chronic remodeling of the ECM network.72,73 Excessive scarring increases the stiffness of the heart, hinders oxygen diffusion, and affects the heart’s diastolic function.
Notably, although the above-described process of AVR is a major factor in the development of HF in patients with MI, ventricular dysfunction may also result from other contributing or superimposed factors. Mechanical complications such as myocardial rupture, septal defects and ventricular free wall rupture, MI complicated by papillary muscle insufficiency, ventricular wall tumors, and septal perforation can aggravate the cardiac load and thus induce and worsen HF. Among these, septal rupture usually has the worst prognosis. 74 In addition, the development of HF after MI is closely related to previous myocardial damage. Long-term chronic myocardial ischemia can cause myocardial fibrosis, which increases the risk of HF after AMI. Patients with repair-induced scars in old infarcts or a history of HF are more likely to redevelop HF after MI, and the combination of other hereditary, metabolic, or toxic injury-related cardiomyopathies may also lead to AVR, LV dysfunction, and HF. Finally, patients who develop worsening of other systemic comorbidities such as anemia, chronic renal failure, and chronic obstructive pulmonary disease are also predisposed to HF after MI. Therefore, the deterioration of heart function in patients with MI and the final progression to HF is a multimechanistic pathway-related process influenced by multiple factors.
HF after MI: Beyond AVR
Although HF can be prevented by the use of reperfusion strategies and cardioprotective drugs as well as the implementation of strategies aimed at preventing LV dysfunction and remodeling, the risk of HF after MI has not been eliminated. One study showed no significant changes in the LVEDV or LV ejection fraction (LVEF) in patients with STEMI during follow-up, but up to 7.5% of patients required hospitalization because of HF during long-term follow-up. 75 In trials comparing colchicine, cyclosporine, and anabolic acid with a placebo, the common endpoint of HF or death still occurred in 13%, 10%, and 26% of patients, respectively, and was not associated with changes in the LVEDV and LVEF despite minimal changes in relative remodeling in patients with STEMI during follow-up.76–78 This suggests that HF after MI can occur without systolic dysfunction and AVR. Additionally, the pathophysiological mechanisms leading to HF may be multifactorial and cannot be explained only by LV systolic dysfunction and ventricular remodeling. These mechanisms may be related to RV dysfunction or HF with a preserved ejection fraction (HFpEF) unrelated to an altered LVEF. 79
HF caused by RV infarction has unique hemodynamic characteristics (Table 1). Although acute proximal occlusion of the right coronary artery presents with typical symptoms of right HF, RV dysfunction may also occur after a larger infarction of the left coronary artery. RV function is an independent predictor of HF development and death in patients with LV dysfunction after MI, and reduced RV systolic function is also a major risk factor for death, sudden death, HF, and stroke after MI.80,81 Compared with the LV, the RV has stronger resistance to permanent infarction. In addition, a series of optimal compensatory measures induced by RV infarction may lead to a greater potential for ischemic myocardial recovery. 82 For example, the RV can maintain its function during moderate coronary artery hypoperfusion, possibly because the increased coronary blood flow from endogenous NO release reduces the myocardial oxygen demand. 83 The ischemic shock of the surviving myocardium usually mediates RV dysfunction. In cases of acute overload, the RV coronary artery perfusion pressure decreases, and the failure of the above compensatory measures leads to severe RV systolic dysfunction. This further reduces the blood flow to the LV, which ultimately manifests as reduced cardiac output and systemic hypotension. Meanwhile, elevated LV end-diastolic pressure reflected in the RV also leads to further expansion of the RV and a decline in systolic function. 84 However, because of the rarity of independent RV infarction, opinions regarding therapy (which are based on animal studies and observational evidence) mainly focus on underlying myocardial ischemia, end-organ perfusion, and potential arrhythmias, advocating that early revascularization may improve short-term and long-term patient outcomes. 85
Differences between RV infarction and LV infarction.
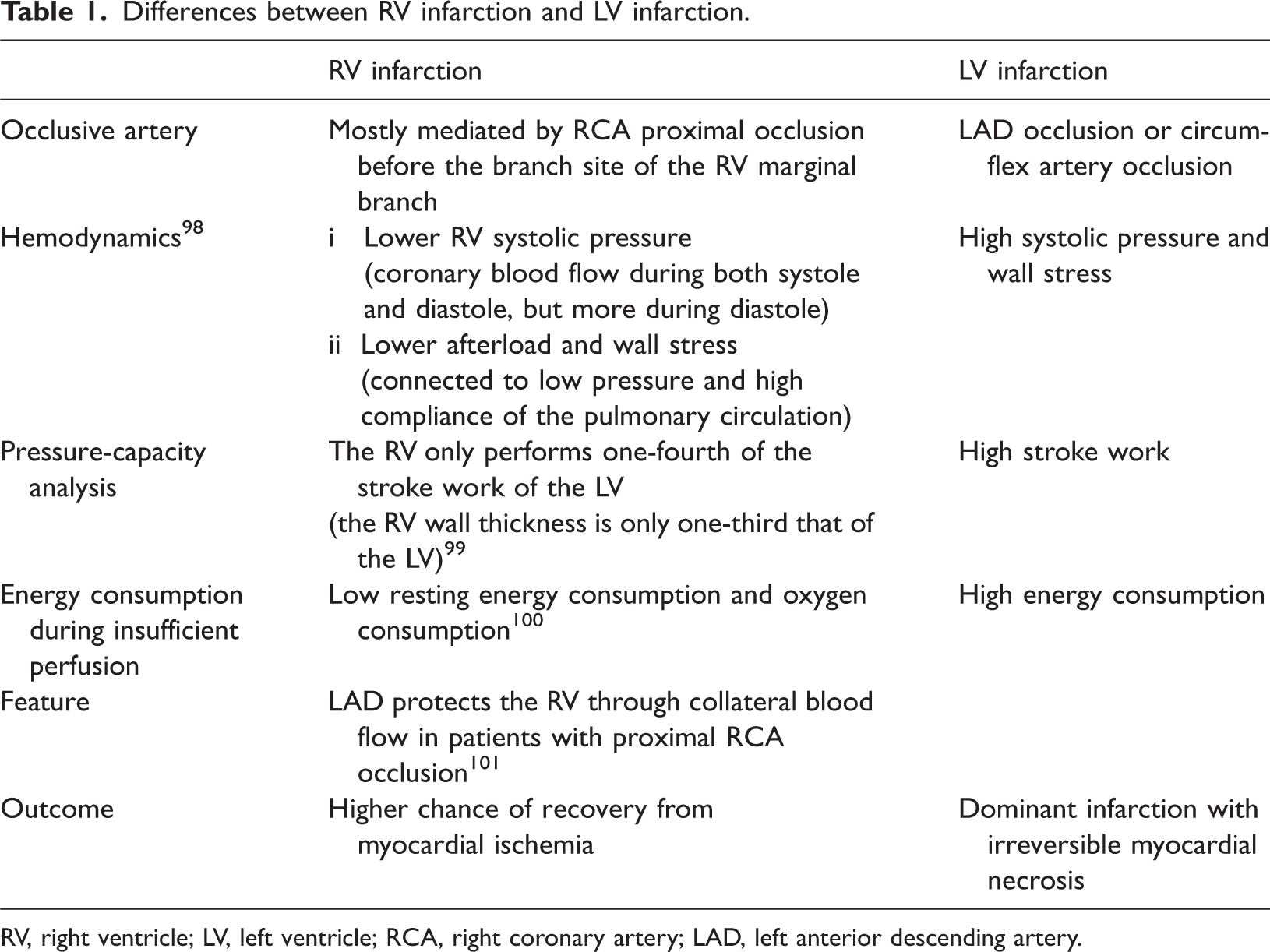
RV, right ventricle; LV, left ventricle; RCA, right coronary artery; LAD, left anterior descending artery.
HFpEF is defined as symptomatic HF with an LVEF of ≥50%. Although the resting systolic function is retained when the LVEF is preserved, patients may still experience symptoms during exercise because of the restricted diastolic reserve, which is affected by increased myocardial stiffness secondary to the elevated LV filling pressure, impaired cardiac output reserve, and persistent chronic ischemia.86–88 Notably, silent MI also has a high incidence rate, although it is asymptomatic. Chronic ischemia may lead to myocardial hibernation and coma as well as a further decline in LV function. Moreover, HFpEF may also result from silent MI because myocardial ischemia is a main factor of HFpEF. 89 In addition, the incidence of HFpEF is strongly associated with hypertension, non-STEMI, and other etiologies such as atrial fibrillation, cardiomyopathy, myocarditis, heart valve disease, and diabetes.80,90
Sex-related differences in prognosis
Women have been found to be at higher risk of new-onset HF after STEMI and to have worse survival rates than men. After correction, although women and men maintain a balance in baseline characteristics and early reperfusion treatment, women with MI still have a higher incidence of HF during hospitalization or after discharge than men. 91 This phenomenon may have three main explanations. First, microcirculatory disorders, which are more common in women, may impair coronary blood flow reserve, making the myocardium more susceptible to ischemia.92,93 Microcirculatory disorders are more prevalent in women than in men, and coronary microcirculatory disease or dysfunction may be a primary cause of angina symptoms in the presence of normal or near-normal coronary arteries. Coronary microvascular dysfunction may also coexist with atherosclerotic coronary artery disease and contribute to the occurrence and development of HF. Second, intrinsic biological differences in the manifestation of some diseases may lead to different prognoses after AMI between the two sexes, such as worse capillary formation and reduced cardiac adaptation to isolated systolic hypertension in women than in men, as well as different responses to treatment between women and men.94,95 Third, other causes of STEMI may have different effects on new-onset HF and mortality after MI in women and men, such as spontaneous coronary artery dissection, plaque erosion, or coronary artery spasm.
Discussion
The development of HF after MI involves cardiac structural and functional changes under neurohormonal regulation, inflammatory immune regulation, and complex pathophysiological interactions between different signaling molecules and pathways. However, despite significant progress in elucidating the underlying mechanisms of HF after MI, many issues remain unresolved. First, the list of molecular factors leading to CM hypertrophy, necrosis, and apoptosis is growing. This makes it important to identify the mechanisms underlying structural and functional alterations in CMs using systematic analytical approaches, such as RNA sequencing and proteomic analysis, which could reveal higher-priority targets for preventing the development of HF after MI. 96 Second, the understanding of the dynamic cardiac matrix profiles in pathological cardiac remodeling remains limited, which may hinder exploration of the mechanisms of cardiac fibrosis to some degree. Therefore, determining the functional domains of ECM components and genetically tracing the myofibroblast lineage will provide an in-depth and comprehensive understanding of the pathogenesis of cardiac fibrosis. This is crucial for developing effective and safe therapies for cardiac fibrosis. 97 However, changes in cellular and tissue components alone may not be suitable as direct targets for HF therapy. Therefore, it is also necessary to develop a better understanding of the cross-regulatory role of neurohormonal system activation and inflammatory immunity in mediating cardiac adaptation and dysfunction after MI. Further investigation of the interactions between different mechanisms may be a future research direction. Although several types of drugs are available to prevent AVR after MI and treat LV dysfunction, individual differences in pathophysiological mechanisms may limit their clinical application. Therefore, the development of innovative drugs will depend on identifying new targets, and cardiac resynchronization therapy is a research hotspot. The gradual clarification of these pathophysiological mechanisms will provide new ideas for different means of treatment to delay the progression of HF after MI and may provide incremental benefits to existing therapies.
Footnotes
Declaration of conflicting interests
The authors declare no conflict of interest in preparing this article.
Ethics
The requirement for ethics approval was waived because of the nature of this study (review).
Funding
This work was supported by the Youth Fund of Ruijin Hospital Affiliated to Shanghai Jiaotong University School of Medicine (No. KYH2022093).