
Abstract
Primary retroperitoneal mucinous cystic neoplasms are rare retroperitoneal tumors, which are histologically similar to mucinous cystic neoplasms of the ovaries. Only 31 cases of primary retroperitoneal mucinous cystic neoplasm with borderline malignancy (PRMCN-BM) have been reported (26 in women and five in men). We describe an additional male patient with PRMCN-BM. A 39-year-old man presented to our hospital with back pain. Twelve years earlier, he had undergone an orchiectomy for a germ cell tumor. Computed tomography showed a 6.9- × 4.4-cm cystic mass in the left pararenal space. Laparoscopic mass excision was performed, and a unilocular cystic mass was found in the pararenal space near the lower pole of the left kidney. A histopathological examination showed a cyst lined by atypical mucinous intestinal epithelium without stromal invasion. Targeted next-generation sequencing identified two hotspot mutations, with one each in the KRAS and GNAS genes. Outpatient follow-up 10 months after surgery showed no evidence of tumor recurrence. PRMCNs are extremely rare retroperitoneal neoplasms, especially in men. These neoplasms are rarely considered in the differential diagnosis of retroperitoneal masses, and their preoperative diagnosis is difficult. Evaluation of additional patients is required to better determine the prognosis of PRMCNs and the optimal postoperative follow-up.
Keywords
Introduction
Primary retroperitoneal mucinous cystic neoplasms (PRMCNs) are rare retroperitoneal tumors that predominantly occur in women. 1 Histologically, PRMCNs resemble ovarian mucinous tumors and are similarly classified into the following three categories: retroperitoneal mucinous cystadenoma, PRMCN with borderline malignancy (PRMCN-BM), and malignant retroperitoneal mucinous cystadenocarcinoma. 2 PRMCN-BM is exceedingly rare, with only 31 (26 in women and 5 in men) cases described in the literature to date. The preoperative diagnosis of PRMCN-BM is challenging, and its pathogenesis, clinical behavior, and management remain unclear because of the small number of registered cases and the absence of pathognomonic clinical and imaging features. 3 The present report describes the sixth case of PRMCN-BM in a male patient and reviews the published data.
Case report
A 39-year-old man presented to our hospital with the complaint of back pain. Twelve years previously, he had undergone an orchiectomy for seminoma at another hospital, and this was followed by four cycles of cisplatin and etoposide chemotherapy. Seven months later, he was transferred to our hospital for follow-up, at which time an abdominal computed tomography (CT) scan was taken. There were no abnormalities, except for the previous orchiectomy site (Figure. 1). He was followed up in our hospital, and there was no recurrence for 12 years.

Abdominal and pelvic computed tomography scan performed after orchiectomy shows no abnormalities in the retroperitoneum.
On the basis of the history of the testicular tumor, the patient underwent blood tests for tumor markers, such as alpha-fetoprotein and β-human chorionic gonadotropin, but the results were normal. An abdominal and pelvic CT scan showed a 6.9- × 4.4-cm cystic mass in the left pararenal space (Figure 2a–c). No mural nodules were found on the mass, and there were no signs of peritoneal implants, ascites, or lymphadenopathy.
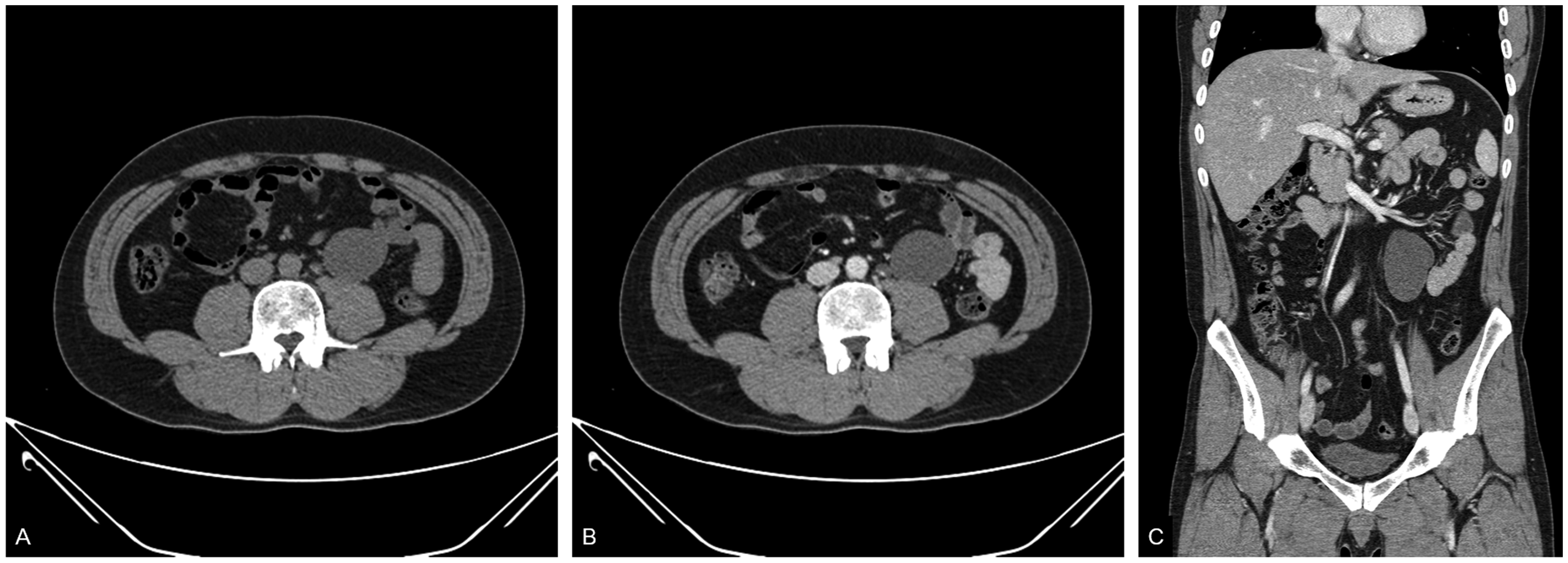
Unenhanced computed tomography scan of the abdomen and pelvis in the axial plane (a) contrast-enhanced images in the axial (b) and coronal planes and (c) show a 6.9- × 4.4-cm homogeneous cystic mass with smooth borders in the left pararenal space.
The neoplastic nature of the mass was uncertain. Therefore, the patient underwent laparoscopic excision of the mass. The mass was located in the pararenal space near the lower pole of the left kidney. After careful dissection of the pararenal fat and surrounding structures, the cyst was extracted gently without spillage of contents. No bag was used for retrieval, and the cyst was not aspirated before extraction. A gross examination of the surgically resected specimen showed a previously disrupted unilocular cyst (5.9 × 4.4 × 3.5 cm) containing mucinous material and evidence of hemorrhage. The inner surface of the cyst was smooth with no mural nodules or papillae. The entire cyst wall was tan-yellow to gray in color. A histopathological examination showed that the cyst wall was lined by atypical mucinous epithelium of the intestinal type, consisting of columnar and goblet cells, without stromal invasion. The cyst wall showed focal calcification, with pools of extravasated mucin. The epithelium displayed slightly atypical proliferation with glandular budding, tufting of the epithelium, and stratification two to three cells thick. The tumor cells displayed mild-to-moderate nuclear atypia without mitoses (Figure 3a). The epithelial lining showed an intestinal phenotype with positive staining for cytokeratin (CK)7, CK20, and CDX2 (Figure 3b–d). Immunohistochemical staining for c-kit and D2-40 was negative. Pathologically, this tumor was similar to mucinous borderline tumors observed in the ovaries. A subsequent thorough workup for any primary tumors failed to identify other lesions in this patient. Therefore, he was diagnosed with a PRMCN-BM.

Histological staining of the tumor sample. (a) Hematoxylin and eosin staining shows the cyst wall with intestinal-type mucinous epithelium and focal nuclear stratification. Papillary tufts with mild cytological atypia can occasionally be seen (original magnification, ×100). (b–d) Staining with antibodies to (b) cytokeratin (CK)7, (c) CK20, and (d) CDX2 shows that the epithelial lining is strongly positive for all three (original magnification, ×100).
The tumor DNA was subjected to targeted next-generation sequencing. Libraries were prepared using Oncomine Comprehensive Assay Plus (Thermo Fisher Scientific, Waltham, MA, USA), and the products were sequenced on the Ion S5 System (Thermo Fisher Scientific). Sequencing data were analyzed using Ion Reporter 5.18. Next-generation sequencing identified two missense mutations, which comprised a c.183A>C (p.Gln61His) missense mutation in exon 3 of the KRAS gene and a c.602G>A (p.Arg201His) missense mutation in exon 8 of the GNAS gene.
We followed the patient with CT examinations at 3 and 10 months after surgery. During this period, the patient remained alive, with no evidence of tumor recurrence.
The reporting of this study conforms to the CARE guidelines. 4
Discussion
PRMCNs are extremely rare retroperitoneal neoplasms, and are histologically similar to mucinous cystic neoplasms of the ovaries. To date, only 31 confirmed cases of PRMCN-BM have been reported (Table 1), with 26 in women and five in men.3,5–24 The first male patient with PRMCN was described in 1994, with only five additional cases of PRMCN-BM in men, including the present patient, reported since this time.7,12,17,18,23
Clinical features of previously reported cases of PRMCN with borderline malignancy

F, female; ND, not described; PRMCN-BM, primary retroperitoneal mucinous cystic neoplasm with borderline malignancy; TR, tumor resection; NED, no evidence of disease; CEA, carcinoembryonic antigen; PRMCN, primary retroperitoneal mucinous cystic neoplasm; M, male; CA 72-4, carbohydrate antigen 72-4; CA 125, cancer antigen 125; CA 19-9, carbohydrate antigen 19-9; CA 15-3, cancer antigen 15-3; Ica, intraepithelial carcinoma; SO, bilateral salpingo-oophorectomy; AFP, alpha fetoprotein; HCG, human chorionic gonadotropin.
Patients with PRMCN-BM range in age from 22 to 68 years, with a median age of 40 years. The clinical presentation of PRMCN-BM is usually non-specific, with no known specific laboratory tests, tumor markers, or radiological findings. Therefore, the preoperative diagnosis of PRMCN-BM is challenging. 25 Serum tumor markers, such as carcinoembryonic antigen, cancer antigen 125, and carbohydrate antigen 19-9, are elevated in a small proportion of these patients, but these elevations do not have diagnostic or prognostic significance. No pathognomonic imaging features of PRMCN-BM have been described to date. These tumors are mainly cystic, with the greatest dimension of the cysts varying from 3 to 28 cm. The cysts may appear unilocular or multilocular, and imaging showed that cysts in only three previously reported cases11,12,31 had a solid component or mural nodule. These tumors are localized almost exclusively to the lateral retroperitoneal spaces, with 9 located in the left side and 11 in the right side. The treatment of choice is complete tumor resection, without cyst rupture, thereby preventing tumor cell dissemination. 25
The prognosis of patients with these tumors remains uncertain because of their rarity and because most patients were not followed up for ≥ 24 months. Outcomes have been reported in 75% of patients with PRMCN-BM who were followed up for a median of 19 months (range, 1–148 months). Based on published case reports, the overall prognosis of patients with PRMCN-BM and documented disease-free survival was excellent, with little evidence of recurrence, metastasis, or cancer-associated deaths, despite one patient with mediastinal lymph node metastases. 6 Therefore, tumor resection is curative, with no need for adjuvant chemotherapy. No consensus has been reached on the postoperative follow-up strategy for patients with PRMCN-BM. However, long-term follow-up data are required to better determine the prognosis of patients with PRMCN.
The pathogenesis of PRMCNs has not been determined because epithelial cells are usually absent from the retroperitoneum.2,10,26 Several hypotheses regarding the origin of these tumors have been proposed. According to one hypothesis, the histological similarity of PRMCNs to ovarian tumors suggests that they arise from ectopic ovarian tissues. However, this hypothesis cannot explain the occurrence of PRMCNs in men or the absence of ovarian tissue from tumors in women. Alternatively, PRMCNs may originate from retroperitoneal monodermal teratomas in which columnar epithelium dominates. In addition, PRMCNs may be remnants of the embryonal urogenital apparatus. The most widely accepted hypothesis is that PRCMNs are derived from invaginated multipotent mesothelial cells that become entrapped in the retroperitoneum during embryonic development, with subsequent mucinous metaplasia and cyst formation. However, in our case, the tumor was not congenital because it was not present on the CT scan performed after surgery (Figure 1).
Targeted next-generation sequencing of tumor DNA in our patient resulted in the identification of two hotspot mutations, with one in KRAS (c.183A>C p. Gln61His) and the other in GNAS (c.602G>A p.Arg201His). No studies to date have determined the molecular profiles of PRMCNs. Therefore, the results in this patient were compared with results in ovarian neoplasms. KRAS mutations have been reported in 30% to 75% of mucinous borderline tumors of the ovaries. 27 In contrast, activating mutations of GNAS occur in only a small percentage (2/29, 6.9%) of mucinous borderline tumors of the ovaries, although they are more frequent in other pre-malignant or non-aggressive mucinous-type tumors of gastrointestinal origin. 27 The co-occurrence of GNAS and KRAS mutations has been reported in only four mucinous ovarian tumors. 27 Additional molecular studies are required to better understand the molecular characteristics of this neoplasm.
Before diagnosing a patient with PRMCN, alternative diagnoses should be excluded by a careful clinical examination, diagnostic imaging, and thorough macroscopic and microscopic evaluations. The differential diagnosis should include metastatic mucinous tumors from the gastrointestinal tract (including the appendix) and the pancreas, which are more common than PRMCN. The possibility of a monodermal variant of a cystic teratoma arising from an undescended testis should also be considered because the retroperitoneum has no epithelial cells.
We describe the rare occurrence of a PRMCN-BM in a male patient. These tumors are rarely included in the differential diagnosis of retroperitoneal masses, and PRMCN-BM is difficult to diagnose preoperatively. The diagnosis of PRMCN-BM involves the exclusion of other considerations, based on careful clinical, imaging, and microscopic evaluations. Surgical excision is the standard approach for the diagnosis and treatment of PRMCN-BM. Evaluation of additional patients is required to better determine the prognosis and optimal postoperative follow-up of this condition.
Footnotes
Author contributions
Seung-Myoung Son made a substantial contribution to writing the manuscript. Chang Gok Woo and Ok-Jun Lee interpreted the pathological data. Seok Jung Yun analyzed the patient’s data. All authors have read and approved the final manuscript.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Ethics statement
This study adhered to the guidelines established by the Declaration of Helsinki and was approved by the Institutional Review Board of Chungbuk National University Hospital (Cheongju, Korea, IRB No: 2022-11-013). The patient consented to publication of the case report.
Funding
This research was supported by the Regional Innovation Strategy (RIS) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (MOE) (2021RIS-001).