
Abstract
Objective
Homologous recombination deficiency (HRD) is the main mechanism of tumorigenesis in some cancers. HRD causes abnormal double-strand break repair, resulting in genomic scars. Some scoring HRD tests have been approved as companion diagnostics of polyadenosine diphosphate-ribose polymerase (PARP) inhibitor treatment. This study aimed to build an HRD prediction model using gene expression data from various cancer types.
Methods
The cancer genome atlas data were used for HRD prediction modeling. A total of 10,567 cases of 33 cancer types were included, and expression data from 5128 out of 20,502 genes were included as predictors. A penalized logistic regression model was chosen as a modeling technique.
Results
The area under the curve of the receiver operating characteristic curve of HRD status prediction was 0.98 for the training set and 0.93 for the test set. The accuracy of HRD status prediction was 0.93 for the training set and 0.88 for the test set.
Conclusions
Our study suggests that the HRD prediction model based on penalized logistic regression using gene expression data can be used to select patients for treatment with PARP inhibitors.
Keywords
Introduction
Homologous recombination repair (HRR) is a DNA-damage repair process that restores DNA double-strand breaks without causing mutations. Double-strand breaks invoke chromosome instability and can cause cancers, and impairments in homologous recombination are seen in some cancers. Homologous recombination deficiency (HRD) is caused by a mutation, deletion, or epigenetic silencing in homologous recombination-related genes including BRCA1/2, CHEK1/2, PALB2, and RAD50. HRD is the main mechanism of tumorigenesis in breast, ovarian, and prostate cancers.1,2
HRD causes abnormal double-strand break repair and results in genomic scars, which are aberrant chromosome integrations including large-scale loss of heterogeneity, 3 telomere allelic imbalance, 4 and large-scale state transitions. 5 These three large-scale aberrant genomic alterations can be quantified by counting,6,7 resulting in the HRD score. 6 Some genomic scar-based HRD tests such as Myriad myChoice CDx or FoundationOne CDx, which involve HRD score calculations, have been approved as companion diagnostics of polyadenosine diphosphate-ribose polymerase (PARP) inhibitor treatment for patients with ovarian cancer8,9 and prostate cancer.10,11
It should be noted that HRD tests based on genomic scars have limitations. 12 As systemic treatment proceeds, various resistance mechanisms are developed and the restoration of HRR by reversion or secondary mutations of HRD-related genes may be possible.13,14 The HRD test cannot reflect HRR restoration because genomic scars make an irreversible footprint and persist even after HRR restoration. 12 Therefore, discordance between the HRD status and sensitivity to platinum or PARP inhibitors may be observed. 15
There is a consensus that HRD assays are necessary to predict PARP inhibitor efficacy, and that an assessment of dynamic tumor evolution other than HRD is important to guide future treatment. 16 In this study, we built an HRD prediction model using gene expression data in various cancer types and applied a previously developed machine learning-based model to predict PARP inhibitor response using gene expression 17 to assess the efficacy of PARP inhibitors in solid tumors.
Materials and Methods
Dataset
The cancer genome atlas (TCGA) data were used for HRD prediction modeling. HRD scores of various TCGA cancer types have previously been published, 18 and were downloaded. The three components of the HRD score are: large (>15 Mb) non-arm-level regions with loss of heterozygosity (LOH), large-scale state transitions (breaks between adjacent segments of >10 Mb), and subtelomeric regions with allelic imbalance. Genome-wide segmentation data for allele-specific copy number and LOH, and tumor purity were estimated using the ABSOLUTE computational method. 19
Normalized gene expression data of the pan-cancer TCGA dataset were estimated from mRNA expression data that were generated using RNAseq and normalized. TCGA gene expression data were downloaded from the National Cancer Institute (NCI) Genomic Data Commons (GDC) website. 20 Ethical approval was not required because public databases were used according to TCGA publication guidelines. 21
Prediction modeling
We constructed an HRD status prediction model using TCGA gene expression data. The target variable was the HRD status, and predictor variables were cancer type and gene expression. Genes with a large median absolute deviation value (greater than third-quartiles) were selected to reduce computational load. The HRD status was defined as positive if the HRD score was ≥42 and negative if it was <42. 6 A penalized logistic regression model was selected as the modeling technique.
Three-quarters of the dataset was designated as the training set and the remainder as the test set according to HRD status stratification. Data preprocessing was performed on the training set using a Yeo–Johnson transformation to resolve skewness, followed by normalization, standardization, and oversampling to resolve the case imbalance with the Synthetic Minority Over-sampling Technique (SMOTE). 22 Data preprocessing was performed using the recipes R package 23 and themis R package. 24
Model selection was performed with a 10-fold validation on a grid of hyper-parameters:
Assessing model performance
The performance of the prediction was estimated using an area under the curve of the receiver operating characteristic curve (AUROC) for HRD status. The modeling process and assessment of model performance were performed using tidymodels 25 and glmnet rpackages. 26
Important predictor selection and gene ontology analysis
Important predictors were selected by coefficients. Genes were selected with a large absolute coefficient value (>0.02). A gene ontology test was undertaken using PANTHER 27 to determine which biological processes are important in HRD status prediction. The following detailed parameters were used: analysis type: PANTHER overrepresentation test (released 2022-07-12); annotation version and release date: PANTHER version 17.0 released 2022-02-22; test type: Fisher’s exact; correction: false discovery rate.
Results
Dataset summary
A total of 10,567 cases of 24 cancer types were included, ranging in number from 45 (cholangiocarcinoma) to 1171 (breast invasive carcinoma). Cancer type abbreviations and the number of cases are listed in the S1 Appendix. Gene expression of 5128 of the 20,502 genes was included as predictors. The number of HRD status-positive cases was 1169 (11.1%). Ovarian cancer showed the highest HRD-positive rate (0.56).
Selecting model and performance estimation
The model with

Hyperparameter tuning and performance assessment in 10-fold cross-validation resampling. The x-axis is a penalty scaling parameter: λ { 10−5, 10−4, 10−3, 10−2, 10−1, 100, color is mixture hyperparameter of penalty function: α {0.0, 0.25, 0.5, 0.75, 1.0}. The y-axis is a mean estimate of the area under the curve of the receiver operating characteristic curve (AUROC) using 10-fold cross-validation resampling.
Performance by cancer type
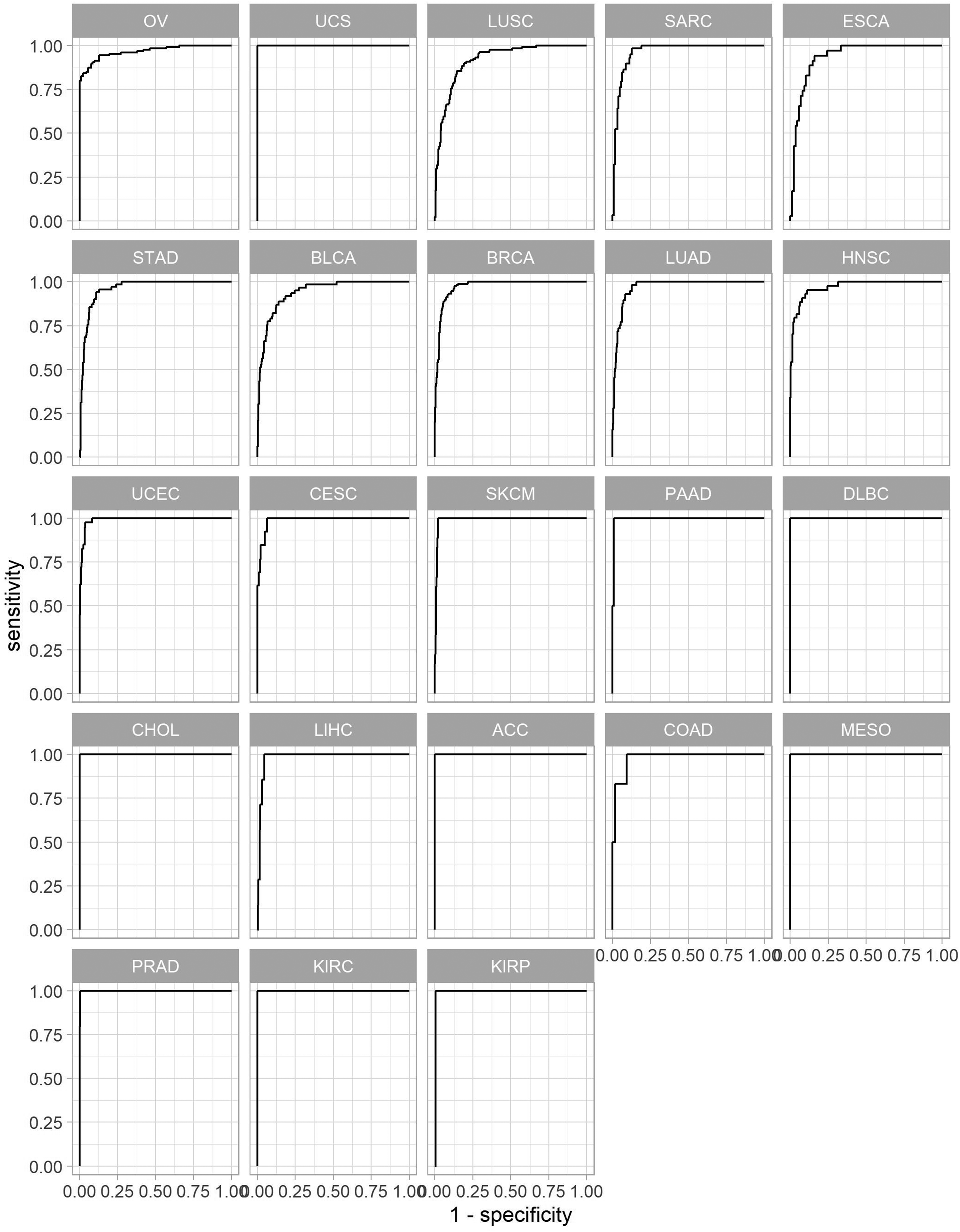
All cancer types showed good prediction in terms of AUROC and accuracy in the training set. In training set prediction, AUROC was above 0.92 for every cancer type and the accuracy was above 0.72 (Table 1 and Figure 2). Most cancer types maintained good performance in the test set prediction, except for uterine carcinosarcoma, lung squamous cell carcinoma, and esophageal carcinoma. In the test set prediction, AUROC was above 0.76 and the accuracy was above 0.74 in every cancer type except uterine carcinosarcoma and pancreatic adenocarcinoma (Table 1 and Figure 3). The cancer types with a low HRD positive rate (<0.10) showed very good accuracy (>0.90) in the test set. Table 1 summarizes the HRD positive rate, accuracy, and AUROC of the training set and test set.
Accuracy and area under the curve of the receiver operating characteristic curve for each cancer type.

Note: Study abbreviations are listed in S1 Appendix.
AUROC, area under the curve of the receiver operating characteristic curve.
Receiver operating characteristic (ROC) curve of homologous recombination deficiency status prediction of the training set.

Receiver operating characteristic (ROC) curve of homologous recombination deficiency status prediction of the test set.
Important predictors and gene ontology test
Genes with large absolute coefficient values can be considered useful in predicting the HRD status. These genes with positive coefficients are described in the S2 Appendix, and those with negative coefficients are described in the S3 Appendix. Genes with large positive coefficients were overrepresented in actin cytoskeleton organization, protein-containing complex subunit organization, the cellular metabolic process, and the organic substance metabolic process. Genes with large negative coefficients were overrepresented in negative regulation of the apoptotic process and the cellular protein metabolic process. Gene ontology analyses of genes with large negative and positive coefficients are described in S4 Appendix.
Discussion
An accumulation of genomic alterations or aberrations based on HRD ultimately leads to the carcinogenesis of solid cancers such as ovary, breast, and prostate cancer. PARP inhibitors such as olaparib and talazoparib inhibit and trap poly(ADP-ribose) polymerase 1, which repairs single-strand DNA breaks (SSB). The prevention of SSB restoration by PARP inhibitors results in double-strand breaks and eventually leads to cellular death in BRCA1/2 mutant or HRD cancers. Based on this mechanism, PARP inhibitors have shown clinical efficacy in BRCA1/2 mutant ovarian, breast, and prostate cancer, and also in HRD-positive ovarian and prostate cancer.
The tumor environment undergoes dynamic evolution as treatment proceeds, so an assessment of the overall tumor biology is warranted. Evaluating genomic characteristics based on gene expression profile can overcome the limitation of not reflecting a change in HRD because the gene expression profile represents the current functional cellular state. Although genomic scars do not disappear once they are created, the HRD gene expression signature changes to the HRR signature if HRR restoration occurs. 28
Our penalized logistic regression model predicted the HRD status with gene expression data in various cancer types with a 0.93 test set AUROC. This prediction performance is similar to the HRD status prediction previously achieved using deleterious BRCA1/2, 29 suggesting that an HRD prediction model based on penalized logistic regression using gene expression data can be applied to patient selection for treatment with PARP inhibitors. This model is preliminary and should be validated in a clinic with prospectively collected cancer tissues, but this study confirms the possibility of HRD prediction using gene expression data. This offers the advantage of evaluating the current HRD status of cancer using serially-collected tumor tissues when disease shows resistance to previous treatment; in contrast, HRD tests based on genomic scars do not reflect dynamic changes in HRD status.
Our prediction model showed good performance in cancer types associated with high HRD-expressing malignancies including ovarian cancer and breast cancer, but poorly predicted other cancer types, including uterine carcinosarcoma and pancreatic adenocarcinoma. This poor prediction performance might reflect underfitting by a small dataset, so a large dataset showing poor prediction may be needed for these cancer types.
Some gene expression-based HRD prediction tests have previously been studied.28,30–32 However, the HRD status in these studies was determined by the presence of pathogenic variants in HRD-related genes such as BRCA1/2. Conversely, our prediction model used an HRD score to determine the HRD status. An HRD test based on genomic scars has the advantage of detecting more patients who can be treated with PARP inhibitors. 9
The primary purpose of this study was to determine whether gene expression data can predict HRD status. However, by investigating important predictors of our model, we also found that genes with large negative coefficients were overrepresented in negative regulation of the apoptotic process. These findings are consistent with previous studies. 33 Apoptosis is suppressed in the response to DNA damage. 34
Our prediction model has some advantages and limitations. The main advantage of HRD prediction with gene expression is that it reflects the current HRD status. To understand how well this predicts the current HRD status, our model should be tested with gene expression profile data collected from patients who showed resistance to PARP inhibitors or platinum. The restoration of HRR is a major mechanism of such resistance, and gene expression datasets from patients showing resistance may reflect current changes in HRD. Additionally, our prediction model was limited with respect to generalization. The dataset used for the modeling process only included 24 common cancer types, so the performance of other cancer types is unknown. Moreover, TCGA RNAseq normalization is cancer set-dependent. Therefore, the standardization of gene expression data processing should be resolved for the practical use of this HRD prediction model using gene expression data. External validation is also needed for practical use.
In summary, our study shows that the HRD prediction model based on penalized logistic regression using gene expression data can be applied to patient selection for treatment with PARP inhibitors. However, gene expression data standardization and validation in cancer with HRD restoration are required to apply these models in clinical testing.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605221133655 - Supplemental material for Prediction of homologous recombination deficiency from cancer gene expression data
Supplemental material, sj-pdf-1-imr-10.1177_03000605221133655 for Prediction of homologous recombination deficiency from cancer gene expression data by Jun Kang, Jieun Lee, Ahwon Lee and Youn Soo Lee in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605221133655 - Supplemental material for Prediction of homologous recombination deficiency from cancer gene expression data
Supplemental material, sj-pdf-2-imr-10.1177_03000605221133655 for Prediction of homologous recombination deficiency from cancer gene expression data by Jun Kang, Jieun Lee, Ahwon Lee and Youn Soo Lee in Journal of International Medical Research
Supplemental Material
sj-pdf-3-imr-10.1177_03000605221133655 - Supplemental material for Prediction of homologous recombination deficiency from cancer gene expression data
Supplemental material, sj-pdf-3-imr-10.1177_03000605221133655 for Prediction of homologous recombination deficiency from cancer gene expression data by Jun Kang, Jieun Lee, Ahwon Lee and Youn Soo Lee in Journal of International Medical Research
Supplemental Material
sj-pdf-4-imr-10.1177_03000605221133655 - Supplemental material for Prediction of homologous recombination deficiency from cancer gene expression data
Supplemental material, sj-pdf-4-imr-10.1177_03000605221133655 for Prediction of homologous recombination deficiency from cancer gene expression data by Jun Kang, Jieun Lee, Ahwon Lee and Youn Soo Lee in Journal of International Medical Research
Footnotes
Data availability
RNAseq data used in this study are publicly available at: https://api.gdc.cancer.gov/data/3586c0da-64d0-4b74-a449-5ff4d9136611. HRD data are available at: ![]() . The code used for data processing is also available in this repository.
. The code used for data processing is also available in this repository.
Declaration of conflicting interests
The Catholic University of Korea, Industry-Academic Cooperation Foundation has been filed a patent for “Modeling method for homologous recombination deficiency predictive model” (Application No. 10-2022-0014718). Jun Kang, Jieun Lee, Ahwon Lee, and Youn Soo Lee are listed as inventors of the patent.
Funding
The authors disclosed receipt (pending publication) of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2021R1I1A1A01043754).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.