
Abstract
β-thalassemia (β-thal) is one of the most prevalent inherited blood disorders in Ganzhou, south China. Next-generation sequencing was used to screen for thalassemia carriers in the general population. During the screening, we identified a novel β-thal variant in a 46-year-old Chinese man, which was validated by Sanger sequencing. Based on the patient’s clinical data, this novel mutation was classified as severe β0. However, the patient was mildly anemic (hemoglobin, 89 g/L), which was inconsistent with typical β0 carrier characteristics. On further evaluation, quantitative PCR indicated the presence of six α genes, while molecular analysis and pedigree analysis revealed the coexistence of αααanti3.7 and αααanti4.2. Therefore, we report a novel β-thal variant combined with six α genes. We describe the patient’s clinical phenotype and the process of molecular diagnosis. This case extends the spectrum of thalassemia variants.
Introduction
The β-globin protein (HBB) is synthesized from the β-globin gene (HBB) located on chromosome 11. β-globin in combination with α-globin forms the most common adult hemoglobin (HbA). 1 Abnormal HBB variants influence every stage of gene expression, affecting the stability and synthesis of the β-globin chain, resulting in inherited forms of β-thalassemia (β-thal). More than 300 variants of HBB resulting in β-thal have been described globally, according to the human hemoglobin (Hb) variant (HbVar) database. 2 HBB variants alter the synthesis of HBB either partially (β+) or completely (β0), with varying effects on clinical severity. 3
β-thal carriers are clinically characterized by a low mean corpuscular volume (MCV) and mean corpuscular Hb (MCH), elevated Hb A2 levels, 4 and normal Hb levels. Common β-thal variants are identified through hematological indices; 17 HBB mutations have been detected in the Chinese population through reverse dot-blot hybridization (RDB). 5 However, rare or novel β-thal variants could be missed when using these common methods. In a population with a high prevalence of thalassemia, couples are at risk of having children with severe β-thal, including those with rare thalassemia. Therefore, gap-PCR, multiplex ligation-dependent probe amplification, and Sanger sequencing are used to identify potential carriers of rare β-thal variants.6,7
The frequency of thalassemia alleles in Ganzhou, southern China, is 9.49% which is relatively high. 8 The Thalassemia Prevention Program plays a vital role in regions where the thalassemia prevalence is high. Thalassemia screening strategies and methods vary among regions. Next-generation sequencing (NGS) is a competitive method that could improve screening for thalassemia carriers, and provide an integrated assessment strategies in populations with a high thalassemia prevalence. 9 To accurately assess the prevalence of thalassemia in the population and avoid missing rare thalassemia variants, NGS was applied for the genetic screening of thalassemia in Ganzhou.
During the screening, a novel heterozygous variant was identified in a 46-year-old Chinese man. This novel frameshift variant in the β-globin gene [NM_00518.4 (HBB):c.194dup and: p.(Lys66Glnfs*8)] was validated through different methods. This β-thal mutation correlated with the abnormal hematological parameters of the patient. We herein report the variant for the first time.
Case presentation
A 46-year-old Chinese man (the patient) and his wife approached our genetic laboratory for thalassemia carrier status screening before their wedding. As shown in Figure 1, this was the patient’s second wedding. Hematological screening of the patient revealed changes in his erythrocyte morphology, low hematological indices, and high levels of hemoglobin (Hb)A2. The patient’s wife had normal hematological indices and HbA2 levels. Hematological data were not available for some family members, including the patient’s 80-year-old mother; his father was deceased. DNA extracted from saliva samples of the patient’s mother showed her genotype to be αααanti3.7/αα, βN/βN. The patient’s ex-wife and 16-year-old daughter refused to participate in the hematological investigation or to provide DNA samples. The patient’s sister and brother agreed to additional family investigation. Study participants gave their informed written consent to provide all hematological and clinical data, and denied any history of blood transfusion. Study approval by an ethics committee or institutional review board was waived because of the voluntary nature of testing.

Pedigree chart and blood cell morphology. (a) Black arrow indicates the patient and (b) Black arrow indicates abnormal red blood cells from the patient seen under optical microscopy.
Full-length HBB, HBA1, and HBA2 sequences were amplified from the DNA of study participants by PCR as previously described by He et al.,
10
and the amplicons were confirmed to contain all exons and introns; this ensured that as many variants as possible could be detected in the HbVar database. Sequencing libraries were constructed using the Illumina HiSeq Sequencing Library Preparation Protocol. These libraries underwent paired-end sequencing for 100 base pairs using an Illumina HiSeq 2000 sequencing system, and bioinformatic analysis was performed as described previously.
10
The direct sequencing of HBB was performed to validate the novel variant using forward primer: 5′-
Family members were also tested for the presence of α- or β-thalassemia-associated common deletions using HBA1 and HBA2 quantitative (q)PCR; cycling conditions and primer details have been described previously. 11 Bands corresponding to αααanti4.2 and αααanti3.7 were obtained using 4% agarose gel electrophoresis following gap-PCR; PCR and primer details have been described previously. 12 Hematological findings of the patient and his wife, sister, and brother are shown in Table 1. The patient’s hematological parameters included a low MCV and MCH, and elevated HbA2 levels. Direct sequencing indicated a HBB frameshift variant (Figure 2); this was a novel variant, HBB:c.194dup (Figure 2). A single G was duplicated at position 194, which resulted in a premature stop codon at codon 72. Prediction tools PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/index.php) were used to predict the impact of the novel variant. According to American College of Medical Genetics and Genomics guidelines, strong evidence of pathogenicity (PVS1, PM2, and PP3) was obtained. The patient did not carry any of the common α-thalassemia variants, as indicated by gap-PCR and NGS. However, his Hb level was low and qPCR showed the amounts of HBA2 and HBA1 to be 4 and 2, respectively (Figure 3). PCR indicated the presence of both αααanti4.2 and αααanti3.7 (Figure 4), so the patient’s thalassemia genotype was described as αααanti4.2/αααanti3.7, βCD64M/βN. The patient’s wife was shown to be αα/αα, βN/βN. The reporting of this study conforms to CARE guidelines. 13
Hematological and molecular data of the family carrying the HBB: c.194 dup mutation.

RBC: red blood cell count; Hb: hemoglobin; MCV: mean corpuscular volume; MCH: mean corpuscular Hb; –: NA; HbA: adult hemoglobin; HbF: fetal hemoglobin.

Sanger sequencing of the HBB: c.194 dup mutation. Forward and reverse sequencing data show the codon 64 duplication in the heterozygous state. Numerous ambiguities are seen as overlapping peaks.
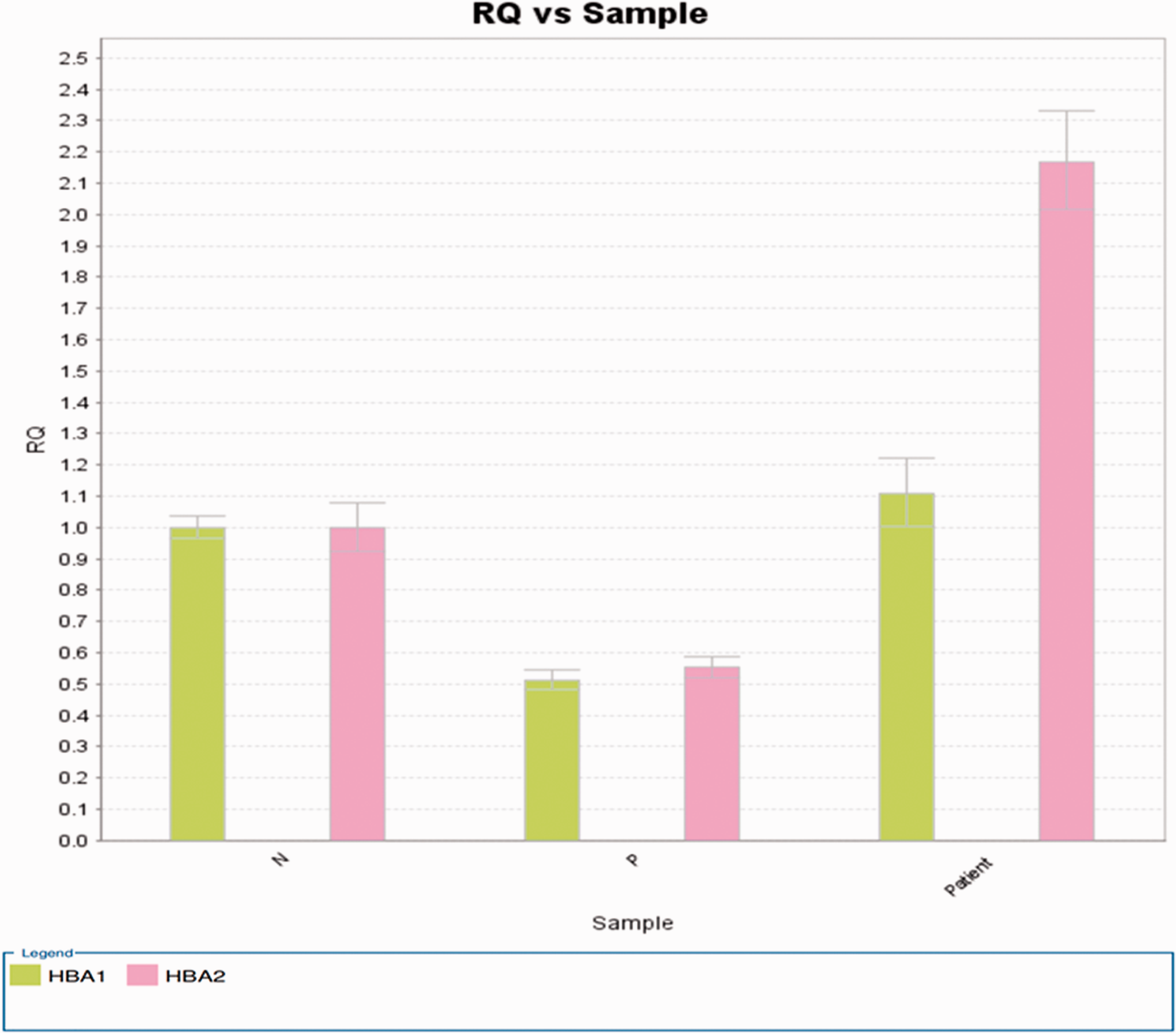
Copy numbers of HBA1 (2) and HBA2 (4) as determined by qPCR.

Agarose gel electrophoresis of PCR amplicons. Lanes 1 and 2, samples from patient’s mother positive for αααanti3.7 and negative for αααanti4.2; lane 3, marker; lanes 4 and 6, negative controls for αααanti3.7 and αααanti4.2, respectively; lanes 5 and 7, positive controls for αααanti3.7 and αααanti4.2, respectively; lanes 8 and 9, samples from patient positive for αααanti3.7 and αααanti4.2 ; lanes 10 and 11, samples from patient’s sister negative for αααanti3.7 and αααanti4.2; lanes 12 and 13, samples from patient’s brother negative for αααanti3.7 and positive for αααanti4.2.
Discussion and conclusions
Many variants responsible for β-thal have been identified and extensively characterized. Most result from single base substitutions, deletions, or insertions in coding regions or at exon–intron classical junction regions that affect almost every stage of HBB expression, including transcription, splicing, polyadenylation, or translation of mRNA. 14
In this study, a novel variant [HBB:c.194dup] was characterized in the heterozygous state. Taking the hematological indices into account, it was expected to be classified as the β0 type. However, the Hb level of the patient was low. Subsequently, qPCR indicated the presence of both αααanti4.2 and αααanti3.7. These triplicated alleles resulted from an unequal crossover between the homologous X-, Y-, and Z-box segments of the α-globin gene cluster during meiosis. Further pedigree analysis ruled out αanti4.2ααanti3.7α/αα as an alternative possibility for the α thalassemia genotype, which could have resulted from an unequal crossover between αααanti3.7 and normal αα.
The patient’s α-thalassemia genotype of αααanti4.2/αααanti3.7 is in agreement with those of his brother and mother. These alpha triplications result in a higher α chain expression in carriers than in healthy individuals. The α chain expression hierarchy is αααanti4.2 > αααanti3.7 > HK αα = αα. 11 Therefore, the coinheritance of triplicated alleles in the patient aggravated the clinical features of the novel heterozygous β0-thal, because of the uneven ratio of α and β chains. This could explain his low Hb levels. However, the Hb level in this patient was much higher than that seen in a previous patient (Hb, 56 g/L) diagnosed with αααanti4.2/αα, β41–42/βN. 11 Additionally, five triplicated α-globin cases coinherited with heterozygote β-thal presented with various clinical manifestations of anemia. 15 Further studies into regulatory regions or genetic modifiers may elucidate the precise effects of this novel variant.
In summary, novel thalassemia variants could be identified in populations where thalassemia is prevalent. Updating the spectrum of variants will enable greater precision in prenatal diagnosis and genetic counselling. Moreover, including new β-thal variants identified locally will improve screening for β-thal mutations using the traditional RDB hybridization method. This will overcome the limitations of regular screening for only the 17 most common β-thal mutations. Future studies should focus on developing a time- and cost-efficient method of comprehensive DNA screening, using tools such as NGS.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605221099013 - Supplemental material for Characterization of a novel HBB:c.194dup variant of the β -globin gene combined with six alpha genes
Supplemental material, sj-pdf-1-imr-10.1177_03000605221099013 for Characterization of a novel HBB:c.194dup variant of the
Footnotes
Acknowledgements
We would like to thank the participant’s family for participating in this study.
Declaration of conflicting interest
The authors have no conflict of interest to report.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding was provided by The Research Project of the Health Department of Jiangxi Province and The Research Project of the Health Department of Ganzhou (grant numbers: 202212360 and 2022-2-218, respectively).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.