
Abstract
Congenital nephrotic syndrome (CNS) is a rare autosomal recessive disorder that occurs in the first 0 to 3 months of life. The course of CNS is progressive, often leading to end-stage renal disease within 2 to 3 years. Most patients with CNS are resistant to glucocorticoids and immunosuppressive drugs. We report a girl aged 1 month and 20 days who was admitted to hospital with a history of abdominal distension and palpebral edema. She was diagnosed with CNS and administered a glucocorticoid (methylprednisolone) for 2 years. Targeted high-throughput next-generation sequencing showed mutations in the NPHS1 gene. She had a favorable outcome after 2 years of treatment. She has remained in complete remission for the last 6 months. From a clinical point of view, the outcome of CNS may be associated with end-stage renal disease or even death. Appropriate pharmacotherapy is beneficial to maintain a normal function and integrity of the glomerular barrier. An aggressive treatment plan is required to save the life of patients with CNS, even if a heterozygous mutation is detected by genetic analysis.
Introduction
Congenital nephrotic syndrome (CNS) can occur in the first 0 to 3 months of life. The most common type of CNS is the Finnish type, which is an autosomal recessive disorder caused by the NPHS1 gene. 1 The NPHS1 gene encodes for nephrin protein, which a fundamental constituent of the slit diaphragm, and plays a crucial role in cell signaling. Mutations of this gene alter the filtration barrier’s function and cause massive protein loss in the urine. 2 As a result, CNS is characterized by massive proteinuria. The course of CNS is progressive, often leading to end-stage renal disease within 2 to 3 years. We report a patient with CNS who was cured by 2 years of glucocorticoid treatment. The reporting of this study conforms to the CARE guidelines. 3
Case report
The patient was a girl aged 1 month and 20 days. She was the first child of a non-consanguineous, 28-year-old, G1P1 mother and a 29-year-old father. The mother was diagnosed with polycystic ovary syndrome 15 years previously, hypertension 6 years previously, and Hashimoto’s thyroiditis 1 month after pregnancy. As a result, the mother took Diane-35 (cyproterone acetate and ethinyl estradiol tablets; Schering GmbH und Co. KG, Weimar, Germany) for 6 years until 1 year before pregnancy, a nifedipine sustained-release tablet for 2 years until pregnancy, and changed to labetalol hydrochloride, while taking Euthyrox (Merck KgAA, Darmstadt, Germany) for 1 year.
The patient was admitted to the hospital with a history of abdominal distension and palpebral edema for more than 10 days. She was born at a gestational age of 37 weeks through a cesarean section, with a birth weight of 3600 g. At delivery, the umbilical cord showed edema, and the placenta was hyperplastic. Nevertheless, the birth weight and height were appropriate for the gestational age. Apgar scores at 1 and 5 minutes were 7 and 8 points, respectively. The placental weight was 1089 g and the index of placental weight/birthweight was 30%.
A physical examination on admission showed a temperature of 36.5°C, a respiratory rate of 40 breaths/minute, and a pulse rate of 120 beats/minute. She also showed abdominal distension, bilateral palpebral edema, and pitting edema in both lower limbs.
A laboratory examination showed massive proteinuria, with a urinary protein of 2+, and a spot urinary protein to creatinine ratio of 28.49, 41.38, and 69.87 mg/mg for three consequent tests in 1 week. There was mild hyperlipemia, with a cholesterol level of 6.1 mmol/L (normal levels, 2.32–5.62 mmol/L), and hypoproteinemia, with an albumin level of 10 g/L (normal levels, 35–55 g/L). Hypocalcemia was also observed (calcium level: 1.95 mmol/L [normal levels, 2.3–2.65 mmol/L]). A blood test showed the following: white blood cell count, 8.39 × 10^9/L; percentage of neutrophils, 32.5%; neutrophil count, 2.7 × 10^9/L; C-reactive protein level, 0 mg/L; and procalcitonin level, 0.9 ng/L. Further laboratory results were negative for the Treponema pallidum particle agglutination assay and toxoplasmosis, rubella, cytomegalovirus, herpes simplex virus, and others. An abdominal ultrasound showed ascites of 3.0 cm. Physical and laboratory results met the diagnostic criteria of nephrotic syndrome.
The placental weight/birthweight index of 30%, massive proteinuria, and hypoalbuminemia without any evidence of infection suggested the diagnosis of CNS. However, there was no family history of this disease. Samples from the patient and her parents were collected for genetic analysis.
Targeted high-throughput next-generation sequencing was performed to screen mutations related to kidney disease in peripheral blood from the patient and her parents. All exons and exon–intron boundaries for NPHS1, NPHS2, CD2AP, PLCE1, and WT1 were analyzed (Table 1 and Figure 1). Mutations were found in the NPHS1 gene. We found c.3312-23C>T in intron 25, c.2207T>C in exon 16, leading to the p.V736A substitution, and c.928G>A in exon 8, leading to the p. D310N substitution. The changes in protein were analyzed by Provean (http: //provean.jcvi.org/seq_submit.php). The cutoff was −2.5, but the PROVEAN scores of V736A and D310N were −3.628 and −4.965, respectively. These mutations were deleterious.
Genetic test results of the patient and her parents.

VUS, variant of unknown significance.
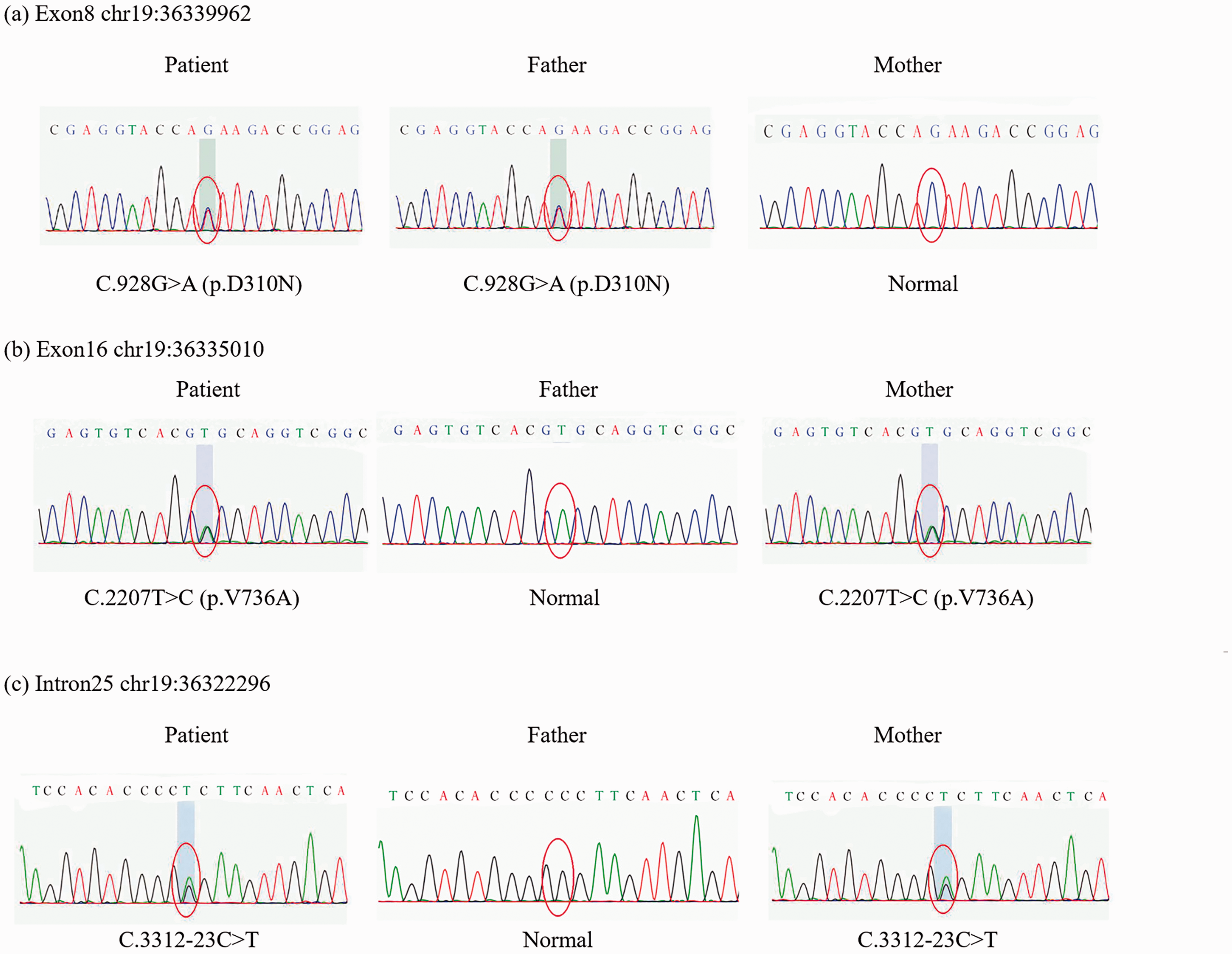
(a) A heterozygous mutation at c.928G > A in exon 8 was inherited from the patient’s father and led to p.D310N substitution. (b) A heterozygous mutation at c.2207T > C in exon 16 was inherited from the patient’s mother and led to p.V736A substitution. (c) The heterozygous mutation c.3312-23C > T in intron 25 was inherited from the patient’s mother.
Because laboratory tests detected massive proteinuria and hypoalbuminemia, glucocorticoid therapy (methylprednisolone, 1.6 mg/kg, once daily) was used to reduce the proteinuria. Intermittent albumin and furosemide injections were initiated to reduce the hypoalbuminemia and edema. Some supportive measures, which comprised taking dipyridamole and vitamin D orally, and infusing dextran, were performed to improve the patient’s symptoms. After 2 months of treatment, the symptoms of proteinuria and hypoalbuminemia began to improve. This regimen was changed to alternate day therapy with methylprednisolone (1.6 mg/kg/day or 1.2 mg/kg/day alternatively), which was reduced to 0.4 mg/kg each month, until the dose was reduced to 1.6 mg/kg every other day. At that time, the symptoms of the patient were considerably alleviated. The methylprednisolone dose was then gradually reduced to 0.2 mg/kg every other day in 1 year, and this low dose was maintained for another 6 months until the patient was in complete remission. The urinary protein became negative, the spot urinary protein to creatinine ratio was 0.33 mg/mg, the albumin level was 35 g/L, and the cholesterol level was 5.62 mmol/L. The details of laboratory tests are shown in Table 2. The patient is currently well and has remained in complete remission for 6 months.
Laboratory test results of the patient.
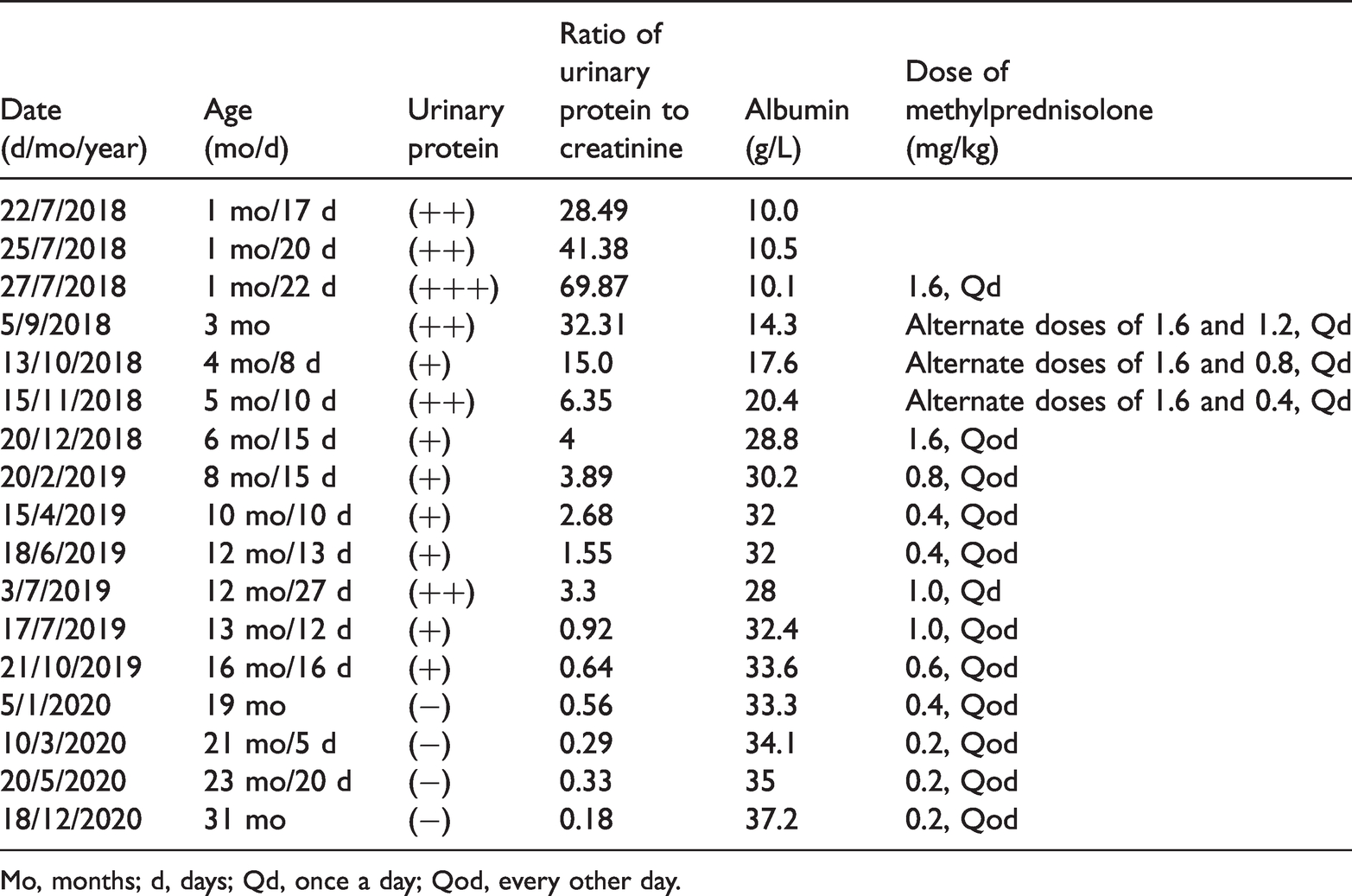
Mo, months; d, days; Qd, once a day; Qod, every other day.
During the whole treatment course, the patient’s CNS relapsed three times because of infection. The patient first relapsed because of an infection on 22 June 2019. The proteinuria was still present even after 10-day antibiotic therapy. At that time, the spot urinary protein to creatinine ratio was 3.3 mg/mg. As a result, the dose of methylprednisolone was increased from 0.4 mg/kg every other day to 1.0 mg/kg every day. After 14 days, the spot urinary protein to creatinine ratio decreased to 0.92 mg/mg. The dose of methylprednisolone was then changed to 1.0 mg/kg every other day. The other two relapses occurred in November 2019 and May 2020. The patient was infected at these times and urinary protein was positive. She was not admitted to hospital. Therefore, we did not change the regime of methylprednisolone or perform any laboratory tests. The patient went into spontaneous remission after taking antibacterial agents in the out-patient department. Fortunately, the dose of methylprednisolone was still appropriate to achieve a sustained benefit without any major adverse reactions.
Discussion
CNS is a hereditary disorder caused by genetic mutations. Most patients with CNS are resistant to glucocorticoids and immunosuppressive drugs because it is not regarded as an immunological disorder. 4 Standard treatment includes renin–angiotensin–aldosterone system inhibitors, albumin infusion, gamma globulin replacement, nutrition with a high-protein, low-salt diet, vitamin and thyroxine substitution, and prevention of infections and thrombotic complications. 5 However, the frequency of intercurrent complications remains high, and the patients’ growth and development are usually retarded. The course of CNS is progressive, often leading to end-stage renal disease or even death. 6
NPHS1 mutation is an important diagnostic criterion of CNS. In our case, the patient showed complete remission of CNS caused by new NPHS1 mutations and she was treated with long-term glucocorticoids. A genetic test showed that the NPHS1 gene mutations were in the loci of chr19-36322296, 36335010, and 36339962. We searched for the NPHS1 gene in ClinVar with the code 602716[MIM], and 469 results showed that none of them contained the above-mentioned loci. Therefore, our patient had three new mutations.
Complete remission of CNS may be rare in many countries and regions, but predicting the outcomes of patients with CNS only depending on NPHS1 mutations is insufficient. 7 A study on Chinese children with sporadic steroid-resistant nephrotic syndrome showed that four patients were in complete remission and responded to prolonged steroid therapy or immunosuppressive agents. 7
Japanese scholars have reported two cases of minimal change lesions in CNS. 8 Both of these patients were cured by 2 mg/kg/day prednisone. On the basis of histopathological lesion patterns, CNS is divided into the four types of diffuse mesangial sclerosis, minimal change disease (MCD), focal segmental glomerulosclerosis, and collapsing glomerulopathy. 9 The terms steroid-sensitive nephrotic syndrome based on clinical features and MCD based on histological features are not entirely equal. However, in clinical practice, especially in children who do not systematically undergo a renal biopsy, these terms are often used as synonyms. 10
A previous study showed that genetic changes in NPHS1 might have a pathogenetic role in some patients with MCD. 11 In our case, the patient showed a new heterozygous mutation genotype and responded to long-term glucocorticoid treatment. The patient’s parents refused a biopsy because she was too young. We could not confirm whether she had MCD. We considered that glucocorticoid treatment was not likely to worsen the patient’s disease if she was resistant to glucocorticoids. Once she was sensitive to glucocorticoids, they could save her life. Renin–angiotensin–aldosterone system inhibitors only provide a palliative treatment. Other treatment, such as nephrectomy or kidney transplantation, would have been a heavy economic burden for the family and unbearable suffering for the child. Even if glucocorticoid treatment had failed, we could have changed to renin–angiotensin–aldosterone system inhibitors in time. Therefore, we decided to perform glucocorticoid therapy in the hope of survival.
From a clinical point of view, the outcome of CNS may be associated with end-stage renal disease or even death. Appropriate pharmacotherapy for CNS is beneficial to maintain the normal function and integrity of the glomerular barrier. However, a more aggressive treatment plan might be required to save the patient’s life, even if a heterozygous mutation has been detected by genetic analysis.
Footnotes
Acknowledgements
The authors would like to thank our patient for allowing her case to be reported.
Author contributions
Mei Han provided conceptual advice and supervised the whole study. Zhong Li and Lanchun Zhuang designed the study and wrote the manuscript. Feng Li collected all data for this case. All authors reviewed the manuscript and agreed to publish it in this journal.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Ethics statement
This article was a case report, and all treatment measures were necessary to save the patient’s life without any research purpose. Therefore, a study protocol was not required. Our institutional ethics committee granted us a waiver of informed consent for publication.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the Dalian Municipality Medical Technological Innovation Project (No. 2019J13SN84).