
Abstract
Objective
This study aimed to clarify the mechanism by which the long non-coding RNA cancer susceptibility candidate 9 (CASC9) alleviates sepsis-related acute kidney injury (S-AKI).
Methods
A lipopolysaccharide (LPS)-induced AKI model was established to simulate S-AKI. HK-2 human renal tubular epithelial cells were treated with LPS to establish an in vitro model, and mice were intraperitoneally injected with LPS to generate an in vivo model. Subsequently, the mRNA expression of inflammatory and antioxidant factors was validated by quantitative reverse transcription polymerase chain reaction (RT-qPCR). Reactive oxygen species (ROS) production was assessed using an assay kit. Apoptosis was detected by western blotting and fluorescence-activated cell sorting.
Results
CASC9 was significantly downregulated in the LPS-induced AKI model. CASC9 attenuated cell inflammation and apoptosis and enhanced the antioxidant capacity of cells. Regarding the mechanism, miR-424-5p was identified as the downstream target of CASC9, and the interaction between CASC9 and miR-424-5p promoted thioredoxin-interacting protein (TXNIP) expression.
Conclusions
CASC9 alleviates LPS-induced AKI in vivo and in vitro, and CASC9 directly targets miR-424-5p and further promotes the expression of TXNIP. We have provided a possible reference strategy for the treatment of S-AKI.
Keywords
Introduction
Sepsis-related acute kidney injury (S-AKI) is one of the most serious complications of sepsis. The symptoms of S-AKI include microvascular dysfunction, inflammation, and metabolic reprogramming, which lead to blood redistribution in the kidneys and acute tubular necrosis. S-AKI is associated with high morbidity and mortality rates, as well as poor prognosis.1,2 However, because of the insufficient understanding of the multifactorial pathogenesis of S-AKI, current treatment strategies focus on supportive and palliative therapy to diminish damage, which is ineffective for curing the disease. Therefore, the development of targeted therapies for S-AKI based on molecular mechanistic research is urgently needed to improve treatment outcomes.
Long non-coding RNAs (lncRNAs) comprise a class of non-protein-coding RNA transcripts more than 200 bp long. They are involved in a variety of biological functions, including cell differentiation, apoptosis, and a multitude of pathophysiological processes. 3 A growing body of evidence has implicated lncRNAs in the progression of a series of cancers.4,5 For example, lncRNAs can mediate the progression of gastrointestinal cancers by regulating autophagy 6 or promote tumor progression by rewiring metabolism. 7 lncRNAs were also found to regulate inflammatory reactions in sepsis-induced organ damage, and they might represent novel diagnostic markers for patients with sepsis.8,9 MicroRNAs (miRNAs) comprise a class of 22-nucleotide single-stranded non-coding RNAs that post-translationally regulate gene expression by promoting the degradation of target mRNAs or inhibiting their translation. 10 Mounting evidence indicates that different miRNAs play diverse regulatory roles in the pathogenesis of S-AKI. 11 The cancer susceptibility candidate 9 (CASC9) gene is located on human chromosome 8q21.11. 12 Previous studies have reported the prognostic indication and regulatory role of this lncRNA in several cancers, including breast cancer, oral squamous cell carcinoma, and gastric cancer.13–15 It is worth noting that CASC9 alleviates S-AKI by regulating the miR-195-5p/pyruvate dehydrogenase kinase 4 (PDK4) axis. 16 Nevertheless, the role of CASC9 in S-AKI remains unclear.
Thioredoxin-interacting protein (TXNIP) is a physiological inhibitor of thioredoxin (TRX)17,18 that functions as a scaffold protein to modulate a number of signal transduction pathways, 19 including inflammatory signals 20 and apoptotic pathways,21,22 TXNIP also induces excessive reactive oxygen species (ROS) production, which causes renal oxidative stress and aggravates the condition of S-AKI. 18 Because of the critical role of TXNIP in tissue damage and inflammatory diseases, tremendous efforts have been invested in the development of TXNIP-targeting therapies for cardiovascular disease and diabetes. 23 Elucidation of the regulatory mechanism of TXNIP in S-AKI might facilitate the development of novel therapies for S-AKI.
Regarding the regulatory relationship between lncRNAs and miRNAs, the competitive endogenous RNA (ceRNA) theory states that different non-coding RNAs compete with each other to regulate downstream targets. For instance, lncRNAs can serve as sponges to competitively bind and adsorb miRNAs, thereby inhibiting downstream target genes.24–26 In this study, we explored the ceRNA regulatory module of CASC9 and miRNAs and the downstream target gene implicated in the regulation of S-AKI. HK-2 human renal tubular epithelial cells were treated with lipopolysaccharide (LPS) to generate an in vitro model to mimic the condition of S-AKI. We found that CASC9 was significantly downregulated in LPS-induced cells. CASC9 overexpression attenuated the expression of inflammatory cytokines and apoptosis induced by LPS and enhanced the antioxidant capacity of cells. In addition, the regulatory role of CASC9 was further evaluated in an in vivo S-AKI mouse model. Overall, our study revealed a functional role of CASC9 in alleviating S-AKI, indicating that targeting the CASC9/miR-424-5p axis might represent a potential intervention strategy for ameliorating S-AKI.
Materials and Methods
Cell culture and transient transfection
HK-2 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in DMEM containing 10% fetal bovine serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 100 U/mL penicillin and 100 mg/mL streptomycin (Invitrogen, Thermo Fisher Scientific) in a humidified cell culture incubator at 37°C with 5% CO2.
HK-2 cells in the log growth phase were trypsinized, washed twice with 1× phosphate-buffered saline (PBS), and then seeded in the corresponding cell culture plates. Subsequently, cells were transfected with overexpression plasmids and/or miR-424-5p mimic using Lipofectamine 2000™ (Invitrogen, Thermo Fisher Scientific) for 24 hours, and quantitative reverse transcription polymerase chain reaction (RT-qPCR) was used to detect the transfection efficiency. miR-424-5p mimic and its negative control, pcDNA3.1-CASC9, pcDNA3.1-TXNIP, and empty pcDNA3.1 vector were obtained from RiboBio Co., Ltd. (Guangzhou, China). Forty-eight hours after transfection, HK-2 cells were then treated with 2 μg/mL LPS27,28 for 24 hours to mimic S-AKI in vitro. The sequences of the miRNA mimic and control were as follows: miR-424-5p mimic, 5′-CAGCAGCAAUUCAUGUUUUGAA-3′, U-3′; miR-NC, 5′-UUCUCCGGGCGUGUCACGUTT-3′.
qPCR analysis
TRIzol (Thermo Fisher Scientific) was used to purify the total RNA of the cells according to the manufacturer’s instructions, and the concentration and purity of the extracted RNA were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Generally, the D260/D280 value was 1.8 to 2.0, indicating high RNA purity. Five micrograms of total RNA were used for reverse transcription using the SuperScript First Strand cDNA System (Invitrogen, Thermo Fisher Scientific). Then, SYBR premix EX TAQ II (Takara, Dalian, China) was used to perform RT-qRCR on the 7500 Real Time PCR System (Applied Biosystems, Thermo Fisher Scientific). The cDNA was used as the template, and the designed primers were used to amplify the target fragment. The 2−ΔΔCt method was used to calculate the relative expression using the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as the internal reference. The primer sequences were synthesized by Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, China) for CASC9 (forward, 5′-TTGGTCAGCCACATTCATGGT-3′; reverse, 5′-AGTGCCAATGACTCTCCAGC-3′), interleukin 1 (IL1; forward, 5′-AGATGCCTGAGATACCCAAAACC-3′; reverse, 5′-CCAAGCACACCCAGTAGTCT-3′), IL6 (forward, 5′-ACTCACCTCTTCAGAACGAATTG-3′; reverse, 5′-CCATCTTTGGAAGGTTCAGGTTG-3′), tumor necrosis factor alpha (TNFα; forward, 5′-GAGGCCAAGCCCTGGTATG-3′; reverse, 5′-CGGGCCGATTGATCTCAGC-3′), superoxide dismutase 1 (SOD1; forward, 5′-GGTGGGCCAAAGGATGAAGAG-3′; reverse, 5′-CCACAAGCCAAACGACTTCC-3′), heme oxygenase (HO)-1 (forward, 5′-CCACAAGCCAAACGACTTCC-3′; reverse, 5′-AAGACTGGGCTCTCCTTGC-3′), TXINP (forward, 5′-TGTGTGAAGTTACTCGTGTCAAA-3′; reverse, 5′-GCAGGTACTCCGAAGTCTGT-3′), GAPDH (forward, 5′-CATGAGAAGTATGACAACAGCCT-3′; reverse, 5′-AGTCCTTCCACGATACCAAAGT-3′), and miR-424-5p (forward, 5′-CAGCAGCAATTCATGT-3′; reverse, 5′-TGGTGTCGTGGAGTCG-3′). For RT-qPCR analysis, three biological samples were measured, each with technical triplicates.
ROS detection
ROS levels were detected using an ROS Assay Kit (ab186027, Abcam, Cambridge, UK) according to the manufacturer’s instructions. In brief, HK-2 cells in the logarithmic growth phase were trypsinized and seeded in 96-well plates at a density of 2000 cells/well. After 24 hours of incubation, cells were treated under the indicated conditions, and cells in different groups were incubated with 100 μL of ROS Working Solution for 1 hour. Finally, cells were subjected to fluorescence detection under a fluorescence microscope with an Ex/Em = 520 nm/605-nm filter. Three independent experiments were performed, and each condition was measured in triplicate.
Western blotting
The cells were lysed with RIPA lysate (Beyotime Biotechnology, Shanghai, China) and centrifuged at 15,000 × g for 20 minutes to remove cell debris at 4°C. The protein concentration was measured using a BAC kit (Thermo Fisher Scientific). Proteins (40 μg) were separated by 10% SDS-PAGE and transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 5% skim milk for 1 hour and incubated with anti-caspase 3 (#9662, 1:1000, Cell Signaling Technology, Danvers, MA, USA), anti-cleaved caspase 3 (#9661, 1:1000, Cell Signaling Technology), and GAPDH (#5174, 1:1000, Cell Signaling Technology) primary antibodies at 4°C overnight. After three washes with TBST, the membrane was further incubated with HRP-labeled secondary antibody (#AB_2099233, 1:2000, Cell Signaling Technology) for 1 hour. Protein bands were developed using an enhanced chemiluminescence kit (Pierce, Thermo Fisher Scientific). ImageJ software (US National Institutes of Health, Bethesda, MD, USA) was used to perform densitometric analysis using GAPDH as the loading control. Error bars represent the standard deviation of three independent experiments.
CCK-8 cell proliferation and viability assay
The CCK-8 assay (Beyotime, Shanghai, China) was used to detect cell proliferation. HK-2 cells were first transfected with pcDNA3.1-CASC9 plasmids or the empty vector for 24 hours. Subsequently, cells were seeded into 96-well plates at a density of 200 cells/well and cultured for 0, 24, 48, or 72 hours in the presence or absence of LPS. To assess the effect of LPS on cell viability, HK-2 cells were treated with different concentrations of LPS (0, 1, 2, 5, and 10 μg/mL) for 24 hours. To perform the CCK-8 assay, 10 μL of CCK-8 solution were added to each well at the indicated time points, and cells were incubated in a humidified cell culture incubator at 37°C with 5% CO2 for 1 hour. The optical density at 450 nm was detected for each well using a microplate reader (BioTek Instruments, Winooski, VT, USA). Three independent experiment were performed, and each condition was triplicated.
Flow cytometry
Cell apoptosis was assessed using an apoptosis kit (BD Biosciences, Franklin Lakes, NJ, USA). In brief, 1 × 106 cells subjected to the indicated treatments were harvested and re-suspended in PBS containing 5% FBS. Cells were stained with 5 μL of Annexin V-PE (BD Biosciences, Franklin Lakes, NJ, USA) and 5 μL of propidine iodide (PI, BD Biosciences) diluted in 500 μL of PBS for 20 minutes at 4°C in the dark. Then, cells were centrifuged, washed three times with PBS, and then re-suspended in 300 μL of PBS. Apoptosis was detected using a BD FACSCanto™ II (BD Biosciences). Three independent experiment were performed, and each condition was triplicated.
Dual-luciferase reporter assay
The interactions of miR-424-5p with CASC9 and TXNIP were analyzed using dual-luciferase reporter assays. The sequences with wide-type (WT) or mutant (MUT) miR-424-5p and CASC9 binding sites were cloned into pmirGLO luciferase reporter plasmid vectors (Promega, Madison, WI, USA) to construct pmirGLO-WT-lncRNA CASC9 and pmirGLO-MUT-CASC9. HK-2 cells were co-transfected with pmirGLO-WT-CASC9 or pmirGLO-MUT-CASC9 plasmids in the presence of miR-NC or miR-424-5p using Lipofectamine 2000 reagent (Invitrogen, Thermo Fisher Scientific). Renilla luciferase (Rluc) plasmids were co-transfected as an internal control. Similarly, pmirGLO-MUT-TXNIP and pmirGLO-MUT-TXNIP plasmids were constructed and transfected into HK-2 cells using the same method. At 48 hours after transfection, the dual-luciferase reporter system (Promega) was used to detect luciferase activity using a microplate reader (BioTek Instruments). The firefly luciferase activity in each reporter was normalized to the Renilla luciferase activity in each sample. Three independent experiment were performed, and each condition was triplicated.
Establishment of the S-AKI mouse model
Thirty male mice (6 weeks old) were randomly divided into three groups (n = 10/group): control group, LPS group, and LPS+CASC9 overexpression group. Mice in the control and LPS groups were injected with CASC9 negative-control adeno-associated virus and those in the LPS+CASC9 group were injected with lncRNA CASC9 overexpression adeno-associated virus (5 × 1012 vg/mouse, 100 µL)29,30 via the tail vein twice a week for 3 consecutive weeks. All adeno-associated viruses were prepared by Genechem Co., Ltd. (Shanghai, China). Three weeks later, mice in the control group were injected with the corresponding volume of PBS, and mice in the LPS group and LPS + lncRNA CASC9 group were intraperitoneally injected with LPS (10 mg/kg)31,32 to induce AKI. Subsequently, blood and kidney tissues were collected from mice after AKI induction. For blood collection, we performed retro-orbital blood collection using a volume of 200 μl. Then, the mice were immediately euthanized using pentobarbital solution (250 mg/kg, i.p.), and finally, the kidney tissue was collected after dissection. Total RNA and protein in kidney tissues and RNA in blood were extracted immediately for subsequent analysis. Kidney tissue samples were used to detect the RNA levels of CASC9, miR-424-5p, SOD1, HO-1, and TXNIP as well as the protein levels of cleaved caspase 3 and pro-caspase 3. Blood samples were used to detect the mRNA levels of IL1, IL6, and TNFα. All experimental procedures were approved by the Laboratory Animal Ethics Committee at the First Hospital of Quanzhou affiliated to Fujian Medical University (No. K-2020033006, approval date: March 30, 2020).
Hematoxylin and eosin (H&E) staining
The kidney tissues of mice in different groups were embedded in paraffin and sectioned into 5-μm slices for H&E staining. H&E staining was performed using an H&E staining kit (ab245880, Abcam). Deparaffinized/hydrated sections were incubated in adequate Hematoxylin, Mayer’s (Lillie’s Modification, Merck, Darmstadt, Germany) to completely cover tissue section for 5 minutes. The sections were rinsed twice in distilled water to remove excess stain. Then, adequate Bluing Reagent (Kehui Biotechnology Co., Ltd. Shanghai, China) was applied to completely cover tissue sections, followed by 30 minutes of incubation. After washing with distilled water, the sections were dehydrated in absolute alcohol, followed by staining with Eosin Y Solution (Leagene Biotechnology Co., Ltd., Beijing, China) by completely covering the tissue for 2 to 3 minutes. The sections were rinsed three times with absolute alcohol and then mounted to a slide, and the images were collected under an inverted microscope (Olympus, Tokyo, Japan).
Statistical analysis
SPSS 13.0 software was used to perform statistical analysis (SPSS Inc., Chicago, IL, USA). All experiments were repeated three times. Data were expressed as the mean ± standard deviation. All quantitative experiments were performed three times. Student’s t-test was used for comparisons between two groups. One-way analysis of variance (ANOVA) was used for comparisons among three groups, and Tukey’s post hoc test was used for pairwise comparisons. Comparisons of data at multiple time points were performed using two-way ANOVA. Statistical significance was indicated by P < 0.05.
Results
CASC9 is downregulated in LPS-treated HK-2 cells
HK-2 cells were treated with LPS in a concentration gradient (0, 1, 2, 5, or 10 μg/mL) for 24 hours. The CCK-8 assay revealed that LPS reduced the viability of cells in a concentration-dependent manner (P < 0.05, Figure 1a). We further treated HK-2 cells with LPS (0 or 2 μg/mL) for 24 hours and used RT-qPCR to detect CASC9 expression. The results indicated that CASC9 expression was significantly decreased by LPS exposure (P < 0.01, Figure 1b), whereas the mRNA levels of inflammatory cytokines such as IL1, IL6, and TNFα and the production of ROS were significantly increased (P < 0.01, Figure 1c and d). By contrast, the mRNA levels of the antioxidant factors HO-1 and SOD1 were significantly reduced following LPS exposure (P < 0.01, Figure 1e). Western blotting was then performed to detect the cleavage of caspase 3, a marker of apoptosis induction. Following LPS exposure, protein level of pro-caspase 3 was reduced, whereas the proportion of cleaved caspase 3 protein was significantly increased (P < 0.001, Figure 1f). These results indicated that the viability and antioxidant capacity of cells were reduced whereas inflammatory responses and apoptosis were increased by LPS treatment. Therefore, LPS could be used to induce AKI-like responses in HK-2 cells, and LPS treatment promotes the downregulation of CASC9 in HK-2 cells.

CASC9 expression is downregulated in an in vitro model of LPS-induced AKI. (a) The human renal tubular epithelial cell line HK-2 was treated with different concentrations of LPS (0, 1, 2, 5, 10 μg/mL) for 24 hours, and cell viability was detected using the CCK-8 assay. (b, c, e) HK-2 cells were treated with 0 or 2 µg/mL LPS for 24 hours. The mRNA levels of CASC9 (b); the inflammatory factors IL1, IL6, and TNFα (c); and the antioxidant factors SOD1 and HO-1 (e) were quantified by RT-qPCR. (d) ROS production was detected using an assay kit. (f) The protein levels of cleaved caspase 3 and pro-caspase 3 were detected by western blotting. Data represent the mean ± standard deviation of three independent experiments. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
CASC9 plays a significant role in LPS-induced AKI
To further explore the functional role of CASC9 in LPS-induced AKI, the pcDNA3.1- CASC9 overexpression plasmid and an empty vector were transfected into HK-2 cells. The overexpression efficiency of lncRNA CASC9 was examined by RT-qPCR, and the results indicated that transfection of the CASC9 overexpression plasmid increased CASC9 levels by 2.5-fold (P < 0.05, Figure 2a). HK-2 cells with or without CASC9 overexpression were treated with LPS for 24 hours. RT-qPCR analysis illustrated that CASC9 overexpression diminished LPS

CASC9 alleviates LPS-induced AKI. HK-2 cells were transfected with CASC9 overexpression or blank control (vector) plasmids and then treated with LPS (2 μg/mL) for 24 hours. (a, b, e) The mRNA levels of CASC9 (a); IL1, IL6, and TNFα (b); and SOD1 and HO-1 (e) in HK-2 cells were detected by RT-qPCR. (c) ROS production was detected using an assay kit. (d) Cell viability was tested using the CCK-8 assay. (f) The proportion of apoptosis cells was detected by flow cytometry. (g) The protein levels of cleaved caspase 3 and pro-caspase 3 were assessed by western blotting. Data represent the mean ± standard deviation of three independent experiments. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
CASC9 acts as an miR-424-5 sponge
To explore the mechanism by which lncRNA CASC9 regulates LPS-induced AKI, we predicted the miRNA targets of lncRNA CASC9 using the LncBase database (http://carolina.imis.athena-innovation.gr/diana_tools/ web/index.php?r=lncbasev2/index-predicted), finding that miR-424-5p was a top-ranked target of CASC9 (Figure 3a). LPS treatment led to miR-424-5p upregulation in HK-2 cells (P < 0.01, Figure 3b). To further explore the role of CASC9 in regulating miR-424-5p, miR-NC/miR-424-5p mimic was transfected into HK-2 cells. The transfection efficiency was examined by RT-qPCR, which revealed a significant increase of miR-424-5p levels after transfection (P < 0.001, Figure 3c). To explore whether CASC9 interacts with miR-424-5p, we performed a dual-luciferase reporter assay using reporter plasmids containing WT or MUT binding sites. We found that miR-424-5p significantly inhibited the luciferase activity of the pmirGLO-WT-CASC9 reporter (P < 0.01), but such an effect was not observed for the pmirGLO-MUT-CASC9 reporter (Figure 3d). In addition, HK-2 cells were transfected with pcDNA3.1-CASC9 plasmids in the presence or absence of miR-424-5p mimic and then treated with LPS. RT-qPCR illustrated that miR-424-5p mimic attenuated the inhibitory effect of CASC9 on ROS production (P < 0.01, Figure 3e), and inflammatory factor expression (P < 0.01, Figure 3f), and caspase 3 cleavage (P < 0.01, Figure 3h) in the presence of LPS. The effect of CASC9 on the upregulation of SOD1 and HO-1 was also compromised by miR-424-5p mimic transfection (P < 0.01, Figure 3g). These results indicate that CASC9 alleviates LPS-induced damage by interacting with miR-424-5p. miR-424-5p might play a role in mediating the beneficial effect of CASC9 on inflammatory responses and apoptosis induction by LPS.

miR-424-5p is competitively inhibited by CASC9 as its downstream target. (a) The target of CASC9 was predicted using a bioinformatics website (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2/index-predicted). (b) miR-424-5p expression in HK-2 cells treated with LPS (0 or 2 µg/mL) was detected by RT-qPCR. (c) miR-NC/miR-424-5p mimic was transfected into cells, and miR-424-5p expression in HK-2 cells was detected by RT-qPCR. (d) The interaction between CASC9 and miR-424-5p was tested using a dual-luciferase reporter assay. (e–h) HK-2 cells were first co-transfected with CASC9 overexpression plasmids in the presence or absence of miR-NC/miR-424-5p mimic and then treated with LPS (2 μg/mL) for 24 hours. (e) ROS production was detected using an assay kit. (f–g) The mRNA levels of IL1, IL6, TNFα, SOD1, and HO-1 were assessed by RT-qPCR. (h) The protein levels of cleaved caspase 3 and pro-caspase 3 were detected by western blotting. Data represent the mean ± standard deviation of three independent experiments. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
TXNIP is a downstream target of miR-424-5p
Previous studies revealed the involvement of TXNIP in the regulation of renal injury during ischemic AKI 33 and tubulointerstitial fibrosis in diabetic kidneys. 34 Based on these findings, we used the Targetscan database (http://www.targetscan.org/vert_72/) to identify the downstream target of miR-424-5p, finding that TXNIP was the consensus target of miR-424-5p (Figure 4a). TXNIP was downregulated in LPS-induced HK-2 cells (P < 0.01, Figure 4b). Then, the pcDNA3.1-TXNIP overexpression plasmid and the empty vector were transfected into the cells. RT-qPCR validated the upregulation of TXNIP after transfection (P < 0.01, Figure 4c). To study the functional interaction between miR-424-5p and TXNIP, a dual-luciferase reporter assay was performed. miR-424-5p significantly inhibited the luciferase activity of pmirGLO-WT-TXNIP reporter (P < 0.01), but no significant inhibition was observed in cells transfected with pmirGLO-mut-TXNIP reporter (Figure 4d). To further investigate the role of TXNIP as a target gene of miR-424-5p in LPS-induced cell damage, HK-2 cells were transfected with miR-424-5p mimic or TXNIP overexpression plasmids alone or in combination. miR-424-5p mimic promoted ROS production, inflammatory factor expression, and caspase 3 cleavage in HK-2 cells treated with LPS (P < 0.01), whereas TXNIP overexpression attenuated the effect of miR-424-5p mimic (P < 0.01, Figure 4e, f, and h). The downregulation of SOD1 and HO-1 induced by miR-424-5p mimic was partially restored by TXNIP (P < 0.001, Figure 4g). Together, these results indicate that TXNIP is a downstream target negatively regulated by miR-424-5p.
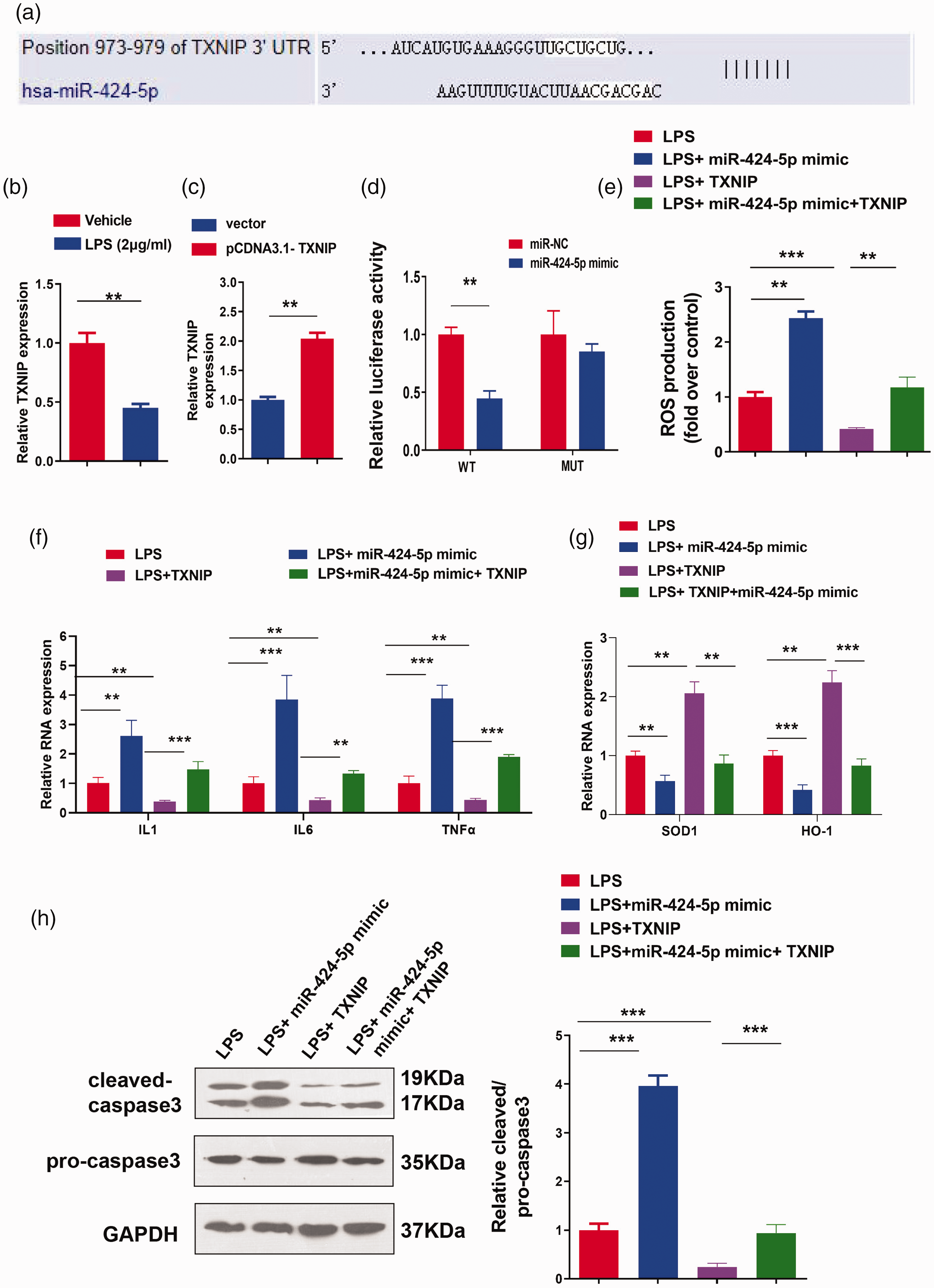
miR-424-5p inhibits the expression of the downstream target molecule TXNIP. (a) The downstream target gene of miR-424-5p was predicted using a bioinformatics websites (http://www.targetscan.org/vert_72/). (b) The mRNA level of TXNIP was assessed by RT-qPCR after HK-2 cells were treated with LPS (0 or 2 µg/mL). (c) pcDNA3.1 empty vector or pcDNA3.1
CASC9 alleviates LPS-induced AKI in vivo
To further verify the functional role of CASC9 in LPS-induced AKI in vivo, mice were injected with 100 μL of CASC9 overexpression adeno-associated virus via the tail vein for 3 weeks, followed by an intraperitoneal injection of LPS to induce infectious AKI. H&E staining illustrated that LPS induced kidney injury in mice, and CASC9 greatly ameliorated this kidney injury (Figure 5a). RT-qPCR analysis demonstrated that compared with the findings in the control group, CASC9 and TXNIP expression was decreased following LPS exposure, whereas that of miR-424-5p was increased (P < 0.01). CASC9 overexpression attenuated these effects of LPS (P < 0.05, Figure 5b). IL1, IL6, and TNFα expression in blood and caspase 3 cleavage in kidney tissues were also increased by LPS treatment (P < 0.01). Consistently, CASC9 overexpression diminished LPS-induced inflammatory factors and caspase-3 cleavage (P < 0.05, Figure 5c and e). In addition, the expression of SOD1 and HO-1 was decreased in mouse kidneys after LPS induction (P < 0.01), and CASC9 overexpression partially rescued their expression (P < 0.05, Figure 5d). Collectively, these results demonstrate that CASC9 overexpression alleviates LPS-induced AKI in an LPS-induced mouse model.
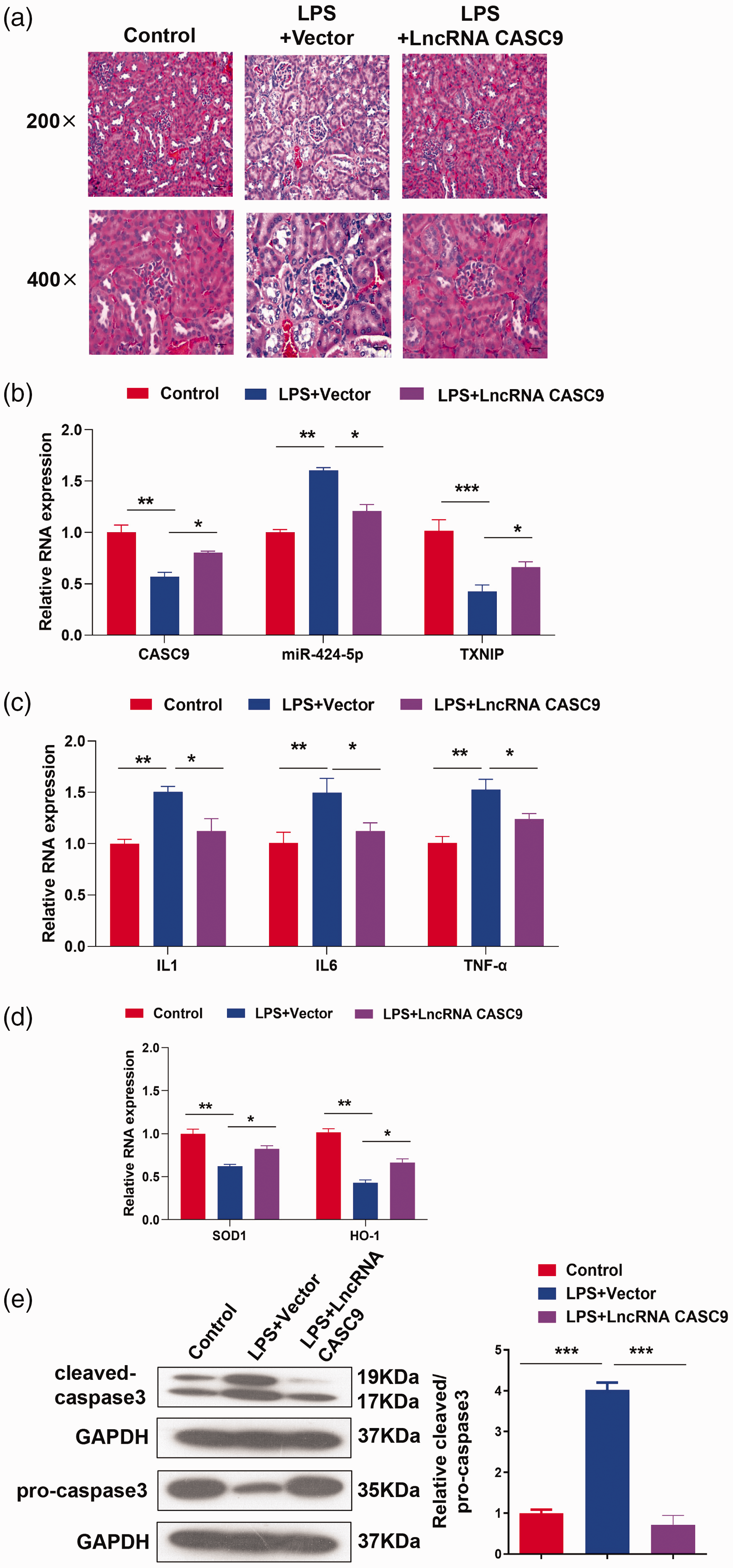
CASC9 alleviates LPS-induced AKI in vivo. CASC9 overexpression adeno-associated virus (100 μL) was injected into the mice through the tail vein. Three weeks later, LPS was injected intraperitoneally (10 mg/kg) to induce infectious AKI in mice. Mice were divided into control, LPS+vector, and LPS+CASC9 groups (n=10 mice/group). (a) The effect of CASC9 on kidney injury was evaluated by H&E staining. Scale bar: 40 μm (×200) or 20 μm (×400). (b) The expression of CASC9, miR-424-5p, and TXNIP in kidney tissue was detected by RT-qPCR. (c) The mRNA levels of IL1, IL6, and TNFα in blood samples were assessed by RT-qPCR. (d) The mRNA levels of SOD1 and HO-1 were detected by RT-qPCR. (e) The protein levels of cleaved caspase 3 and caspase 3 were detected by western blotting. Data represent the mean ± standard deviation of three independent experiments. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Discussion
A number of lncRNAs have been implicated in tissue damage responses. A previous study found that the lncRNA CRNDE was differentially expressed in colorectal neoplasia, and it could protect renal cells against sepsis-induced kidney injury by regulating the miR-181a-5p/peroxisome proliferator-activated receptor α pathway. 35 Depletion of the lncRNA metastasis-associated long adenocarcinoma transcript 1 induces inflammatory responses against sepsis by upregulating miR-150-5p to modulate the nuclear factor-kappaB signaling pathway. 36 Taurine-upregulated gene 1 alleviates sepsis-induced AKI by targeting miR-34b-5p/GRB2-associated-binding protein 1. 37 CASC9 relieves sepsis-induced AKI by regulating the miR-195-5p/PDK4 axis. 16 The aforementioned studies indicated that lncRNAs play crucial roles in the response to sepsis-induced organ damage. In our study, we found that CASC9 can alleviate S-AKI in vitro and in vivo. CASC9 was downregulated in LPS-induced AKI cell and mouse models. We also found that CASC9 overexpression inhibited inflammatory factor expression and apoptosis induced by LPS and promoted SOD1 and HO-1 expression. Together, these data suggest that CASC9 has a protective role to mitigate damage induced by LPS in AKI.
Emerging evidence suggests that a large number of miRNAs are implicated in the pathogenesis of AKI, supporting their potential as for diagnosing AKI. Detailed investigation into the molecular mechanisms will both provide new insights into AKI pathogenesis and renal repair and facilitate the development of new strategies for the diagnosis and treatment. 38 Previous studies illustrated that miR-93-5p can be packaged in extracellular vesicles and secreted by endothelial progenitor cells to alleviate sepsis-induced AKI. 11 miR-424-5p also plays an important role in muscle development 39 and tumor progression in cancers including papillary thyroid cancer, non-small-cell lung cancer, and cervical cancer,40–42 but its role in S-AKI remains to be elucidated. Our study suggests that miR-424-5p is a target of CASC9, and the interaction between miR-424-5p and CASC9 is involved in regulating LPS-induced cell damage.
A previous study indicated that the mitochondrial ROS/TXNIP axis promotes kidney damage induced by ischemic AKI through activating the NOD-like receptor protein 3 inflammasome. 33 In contrast, forkhead box O1 can reduce oxidative damage by regulating the TXNIP/TRX axis in diabetic kidneys. 34 These findings indicate that TXNIP participates in renal oxidative stress and regulates AKI. 18 Interestingly, a recent report demonstrated that miR-30c-5p alleviated AKI by targeting TXNIP. 43 In this study, TXNIP appeared to function as a pro-inflammatory protein that induces tissue or cell inflammation and activates autophagy and apoptosis. Conversely, our study suggested that elevated TXNIP expression reduced kidney damage induced by LPS. In accordance with our finding, Park et al. revealed that deletion of TXNIP enhanced hepatic steatosis, inflammation, and fibrosis, accompanied by impaired autophagy and fatty acid oxidation (FAO) in methionine choline-deficient diet-fed mice. Mechanistically, TXNIP directly interacts with phosphorylated-protein kinase AMP-activated catalytic subunit alpha (PRKAA), leading to the inactivation of rapamycin kinase complex 1 and inhibition of nuclear translocation of transcription factor EB, thereby augmenting autophagy. These findings suggest that elevated TXNIP expression ameliorates steatohepatitis by interacting with PRKAA to maintain autophagy and FAO. Targeting TXNIP may be a potential therapeutic approach for nonalcoholic steatohepatitis. 44 The reported anti-inflammatory and anti-fibrosis functions of TXNIP are consistent with our finding that TXNIP inhibits LPS-induced kidney injury.
Our studies found that TXNIP is a target gene of miR-424-5p, and the miR-424-5p/TXNIP axis mediates the regulation of S-AKI by CASC9. We uncovered a new mechanism by which CASC9 alleviates S-AKI. We found that CASC9 acts as a sponge to adsorb miR-424-5p, thereby releasing its inhibition of TXNIP. Targeting miR-424-5p, TXNIP, or the combined miR-424-5p/TXNIP axis might represent possible approaches to treat S-AKI in the future. In addition, CASC9 and miR-424-5p may be used as novel diagnostic markers for S-AKI, although the feasibility of using these molecules as biomarkers or prognostic indications requires further validation.
Our study also had certain limitations. Although we verified the role of the CASC9/miR-424-5p/TXNIP axis in LPS-induced AKI in HK-2 cells, we only demonstrated the alleviating effect of CASC9 on LPS-induced AKI in mice. The roles of mir-424-5p and TXNIP in LPS-induced AKI in mice require further investigation. In addition, the role of the CASC9/miR-424-5p/TXNIP axis must be verified in additional cell lines.
In conclusion, we unveiled an important role of CASC9 in regulating LPS-induced AKI and the associated mechanism. CASC9 significantly alleviated the symptoms of LPS-induced AKI in vivo. CASC9 overexpression also inhibited inflammation and apoptosis and enhanced the antioxidant capacity of cells in vitro. In terms of the mechanism, we found that the miR-424-5p/TXNIP axis is a crucial regulatory module for CASC9 in LPS-induced AKI. CASC9 maintains TXNIP expression through competitive inhibition of miR-424-5p.
Footnotes
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Authors’ contributions
HPF participated in the study design, performed experiments, analyzed and interpreted data, and wrote the manuscript. ZXZ and JJX participated in the study design and performed experiments. YTL and CWG and HY contributed to acquisition of data and revised the manuscript critically for important intellectual content. HY conceived the study, interpreted data, and wrote the manuscript. All authors read and approved the final manuscript.
Data availability statement
Data are available upon reasonable request.